(1)
Asheville, NC, USA
The year 2017 will mark the 200th anniversary of the 1817 publication of James Parkinson’s monograph “An Essay on the Shaking Palsy.” In his monograph, Parkinson described six individuals suffering from a condition involving a bent posture, involuntary tremulous motion and weakened muscular action. Today, Parkinson’s disease (PD) in its various forms is recognized as the second most prevalent neurodegenerative disorder. It has an incidence rate of 1 % for individuals age 65 and older and that rate increases to 5 % for people past the age of 85.
There are six major clinical symptoms of the syndrome known as parkinsonism . Three of these are early signs of the “shaking palsy.” These are: tremor at rest, rigidity, and slowness of movement (bradykinesia). The remaining three signs appear later as the condition worsens. These signs are: postural abnormalities (bent posture), postural instabilities, and freezing of gait often resulting in falls. These are the key clinical indications of the syndrome, and as such were described in the classical textbooks on neurology by Gowers (1893), Oppenheim (1911), and Wechsler (1932).
Parkinson’s disease, the chief form of parkinsonism , begins in a specific region of the midbrain known as the substantia nigra pars compacta (SNc) and from there spreads to other brain region. The association of the disorder with specific anatomical regions took many years to establish. First, researchers in the late 1800s had found that in the basal ganglia and substantia nigra were affected in the disease. The latter region is enriched in the dark pigment neuromelanin, hence its name, and undergoes depigmentation when neurons are lost. The connection to the SNc was strengthened by Lewy and by Konstantin Tretiakoff (1892–1958), who in his 1919 doctoral dissertation identified the SNc as the primary starting point of the disease and named the Lewy bodies , the chief histopathological sign of PD, after Lewy.
SNc neurons use dopamine as their neurotransmitter, and project their axons to neurons in the striatum responsible for initiating and controlling movement. The lack of adequate dopamine signaling results in failed motor control, and leads in the later stages of the disease to cell death and dementia. A landmark event in treating Parkinson’s disease was the discovery in 1957 by Arvid Carlsson that l-dopa could reverse the symptoms of parkinsonism . Up until that time, depletion of serotonin was thought to be responsible for the observed symptoms, but that idea was shown to be incorrect by Carlsson. His discovery was followed shortly thereafter in 1960 by findings by Herbert Ehringer and Oleh Hornykiewicz that dopamine levels were depleted in the striatum of humans suffering from parkinsonism. That discovery established for the first time the association of an abnormality in a particular chemical occurring in a specific region of the brain with a neurological disorder. That discovery led to the initiation by Hornykiewicz of clinical trials with levodopa in 1961 and by the mid-1970s that treatment had become the accepted approach for reducing the symptoms of parkinsonism.
For many years it was felt that PD lacked a genetic component. Instead, environmental factors leading to oxidative stress were thought to be responsible for the disease. Support for this belief was provided by the discovery in 1983 that the chemical agent 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can produce signs in neurons closely resembling those of parkinsonism . This chemical agent can readily pass through the blood–brain barrier (BBB); it is oxidized to a highly toxic MPP+ form by the enzyme monoamine oxidase B (MAO-B), and is taken up by the dopamine transporter. Once in the neurons it (1) inhibits complex I of the mitochondrial electron transport chain (ETC), (2) impairs respiration, (3) increases production of superoxide, and (4) disrupts calcium homeostasis and associated cellular functions. In a further development of an environmental factor theme, the widely used pesticide rotenone was found to be an even better trigger of parkinsonism-like symptoms in mammalian neurons. Here too the main site of action of the agent was mitochondrial complex I. And a few years later dopaminergic neurons were found to be vulnerable to the herbicide paraquat.
Evidence keeps accumulating pointing to the involvement of environmental toxin s including heavy metals in the etiology of Parkinson’s disease. One finding that is consistent across almost all studies is the generation of oxidative stress , that is, the toxins damage components of the mitochondrial ETC resulting in increased reactive oxygen species (ROS) and reactive nitrogen species (RNS) production (Table 9.1). These toxins are widely used in the laboratory today to reproduce features of PD in test subjects. Using these experimental models, researchers can dissect the steps in the pathogenesis of the disease and test possible therapeutic drugs.
Table 9.1
Environmental chemicals and other toxins widely used to model Parkinson’s disease
Toxins | Description |
|---|---|
MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine generates parkinsonism through the actions of its metabolite MPP+. It is lipophilic and readily passes through the BBB. It damages mitochondrial complex I |
CCCP | Carbonyl cyanide m-chlorophenylhydrazone is a mitochondrial membrane depolarizer. It inhibits oxidative phosphorylation and triggers mitophagy |
Rotenone | Lipophilic insecticide that readily passes through the BBB. Damages mitochondrial complex I. Activates microglia. Generates α-synuclein-positive Lewy bodies |
Paraquat | Herbicide that undergoes channel-mediated passage through the BBB. Damages mitochondrial complex I. When combined with maneb generates a form of PD comparable in magnitude to MPTP-induced forms |
Maneb | Maneb, a fungicide, utilizes manganese ethylene-bis-dithiocarbamate (Mn-EBDC) as its main active ingredient. Damages mitochondrial complex III |
6-OHDA | 6-Hydroxydopanine is used in the laboratory to create a PD model that can be used in drug development. It cannot pass the BBB but can be administered directly through injection. It impairs mitochondrial complex I |
There are over one million Parkinson’s disease sufferers in the United States and this neurological disorder is second only to Alzheimer’s disease in its frequency of occurrence. The focus on environmental factors for its cause began to shift in 1997 with two sets of findings. First, Polymeropoulos discovered that the appearance of PD in certain individuals was associated with mutations in α-synuclein, a small, 140-amino-acid-residue protein enriched in the presynaptic terminals of neurons. Second, Spillantini established that the same protein, α-synuclein, was the main component of the Lewy bodies , the postmortem defining feature of Parkinson’s disease and dementia with Lewy bodies.
Three PD-causing mutations in SNCA, the gene that encodes α-synuclein, were found. The first, the 1997 discovery, involved the substitution of threonine for alanine at position 53 (A53T). The second, found within the next year, was that of a proline substitution for alanine at position 30 (A30P) and the third in 2004 was that of E46K. In just the last few years, three more PD-associated mutations in the SNCA gene have been identified. These mutations, namely H50Q, G51D and A53E, are clustered in a narrow region spanned by the previously identified E46K and A53T mutations. These mutations occur in families with a history of developing PD in an autosomal dominant fashion. By altering α-synuclein’s conformation the mutations influence its ability to oligomerize and fibrillize. They also produce changes in its subcellular localization and alter its function. Further reinforcement of the connection between PD and α-synuclein came from multiple experimental studies showing that loss of α-synuclein results in failures in the nigrostriatal dopamine system and leads to neurodegeneration.
α-Synuclein expression levels are important. The connection between concentration and disease was firmly established by the discovery in 2003 of a triplication in the SNCA gene of Parkinson’s disease sufferers. It was reinforced the following year when a duplication in SNCA was shown to cause PD. In another, large-scale genetic study, polymorphisms in the SNCA promoter were uncovered. These three studies firmly establish that as the expression level of α-synuclein increases so does the risk for developing PD. This theme is a familiar one having been encountered and discussed at length with regard to amyloid-β generation and clearance in the last chapter.
9.1 Multiple Pathways to Parkinson’s Disease
This chapter begins with an examination of what makes the substantia nigra neurons so special that they and they alone are the locus of the disease onset. These neurons possess a number of striking features centered on their unusual morphology and unique biophysical properties. These properties are examined along with an overview of the circuitry involved in PD. Alpha-synuclein is then examined. Since its discovery a growing list of PD-associated mutated genes has been uncovered. The most prominent and best studied of these genes and the proteins they encode are listed in Table 9.2. As noted in the table leucine-rich repeat kinase 2 (LRRK2), like α-synuclein, is inherited in an autosomal dominant fashion, while the remaining risk factor s are inherited in an autosomal recessive manner. Significantly, three of these risk factors—phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1), parkin, and DJ-1 function in protein quality control pathways. Leucine-rich repeat kinase 2 (LRRK2) operates as a lipid-raft/caveolae -associated signaling platform, and it too has a prominent role in protein quality control.
Table 9.2
Key genetic factor s implicated in the pathogenesis of Parkinson’s disease
Locus | Protein | Description |
|---|---|---|
PARK1—4q21 | α-Synuclein | Presynaptic proteins; AD |
PARK2—6q26-27 | Parkin | E3 ubiquitin ligase; AR |
PARK6—1p35-36 | PINK1 | Mitochondrial serine/threonine kinase; AR |
PARK7—1p36 | DJ-1 | Protects against oxidative stress; AR |
PARK8—12p11.2 | LRRK2 | Mixed lineage kinase; AD |
Lewy bodies are spherically shaped, 5–25 μm in diameter, structures. They contain a dense core of filamentous and granular material surrounded by a filamentous halo. They are composed primarily of neurofilaments, ubiquitin, heat shock protein s , and α-synuclein. These structures are present in almost all sporadic or idiopathic cases of PD that comprise the vast majority of the cases of PD. They are present in most instances of LRRK2-associated PD and in all SNCA-associated PD instances. However, they are largely absent in PINK1 and parkin-associated PD, and their presence or absence in DJ-1 associated forms is unknown.
In Alzheimer’s disease, the various risk factor s all seemed to involve the β-amyloid peptide in one manner or another. They either had a role in its generation, or alternatively, in its clearance. Thus, a single pathway appears at the causal center of the disease. It is already clear from the selective presence or absence of Lewy bodies that PD is a more complex phenomenon. PINK1 and parkin do define a clear pathway, one that involves mitochondrial quality control. That pathway is examined in this chapter along with DJ-1, which resides in a parallel mitochondrial-protective pathway to that of PINK1/parkin. The placement of α-synuclein and LRRK2 in a single pathway is a more challenging endeavor. One place where they do come together is in their mutual impairment of chaperone-mediated autophagy , and that locus of commonality is looked before the mitochondrial pathways are discussed.
Lastly, the risk factor s listed in Table 9.2 are not the only ones that have been found. Instead, there is a growing list of recently discovered genetic risk factors. Some of these point to yet another pathway to PD, one that centrally involves the lysosome and its dysfunction due to mutations in its enzymes and its non-enzymatic transport proteins. These emerging risk factors are discussed towards the end of the chapter. The chapter then ends with an overview of a way that cells dispose of potentially dangerous material when all other means are blocked—they release the materials into the extracellular spaces.
9.2 Nigrostriatal Neurons Have a Number of Special Properties
Recall that in the pioneering 1952 study by Hodgkin and Huxley, two types of voltage-dependent ion channels were identified. First, the opening of sodium channel allowed Na+ ions to enter the neuron resulting in a decrease in the net negative charge of the neural interior, thereby depolarizing the plasma membrane. That was followed after a short delay by the opening of potassium ion channels resulting in an efflux of K+ ions and the repolarization of the membrane. The dual actions involving the opening and closing of sodium and potassium channels produced an action potential that propagated down the squid giant axon.
Central nervous system (CNS) neurons have far more complex signaling, communication and control tasks to perform than that of the squid giant axon and their action potential s reflect the varying requirements. Shown in Fig. 9.1 are three examples of different action potentials and accompanying firing patterns—bursting, tonic, and rhythmic/pacemaking—exhibited by CNS neurons. The repertoire of voltage-gated ion channels possessed by these neurons is quite impressive and underlies their ability to produce the different firing patterns. In particular, there are several dozen different kinds of voltage-dependent potassium channels including some that are dependent on the intracellular calcium concentration in their immediate vicinity. Also, there are also a large number of voltage-dependent calcium channels that allow Ca2+ ions to enter the neurons.
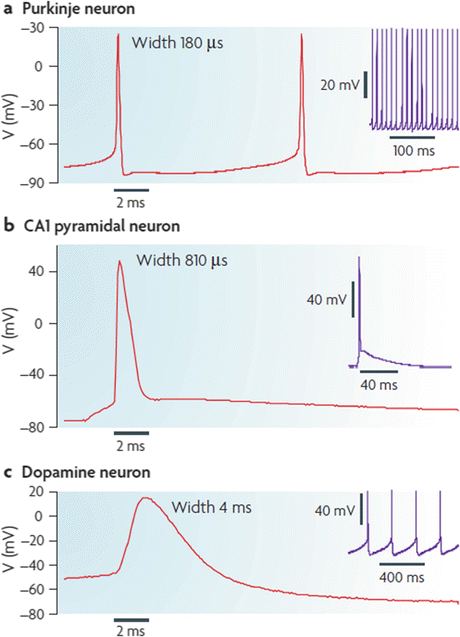
Fig. 9.1
Action potential shapes and firing patterns. (a) Burst firing by cerebellar Purkinje neurons; (b) Tonic firing of hippocampal CA1 pyramidal neurons; (c) Pacemaker firing by SNc dopaminergic neurons (from Bean Nat. Rev. Neurosci. 8: 451 © 2007 Reprinted by permission from Macmillan Publishers Ltd)
The elevation of calcium levels in neurons has multiple effects and its concentration is tightly regulated. Under normal and quiescent conditions, its intracellular concentration is kept some three orders of magnitude lower than its extracellular concentration. Any transient increase in concentration is handled rapidly by an assortment of channels, pumps, and other calcium transporters. These either send the calcium ions back out to the extracellular spaces or into intracellular storage locations. As discussed in the last chapter, the calcium handling system localizes the Ca2+ increases both in space and time, thereby enabling the ion to function as a signaling intermediary, referred to as a second messenger. For example, in response to the arrival of an action potential at the presynaptic terminal Ca2+ enters through voltage-gated calcium channels. The Ca2+ ions diffuse to calcium sensors located on the outer surface of the neurotransmitter-loaded vesicles. This arrival activates effector proteins that trigger vesicle membrane fusion and release of the neurotransmitters into the synaptic cleft.
Substantia nigra pars compacta neurons have a number of special properties that create stressed conditions. These properties render the neurons vulnerable to age-related decline of proteostasis and to buildups in misfolded proteins especially those identified in Table 9.1. SNc neurons:
Possess highly ramified, poorly myelinated axons. Unlike most other neurons their axons form dense axonal bushes. This architecture provides every DA-secreting neuron with a large number of DA releasing sites that can influences tens of thousands of striatal neurons.
Function as autonomous pacemakers. SNc neurons spike on their own in the absence of synaptic input. This enables the neurons to continuously supply dopamine to the recipient striatal neurons. In order to spike continuously in that manner, the neurons exhibit rather depolarized membrane conditions typical of pacemakers. They have a high metabolic burden and rely heavily on oxidative phosphorylation to meet their metabolic needs.
Have weak calcium handling capabilities: Calcium concentrations are tightly regulated in most cells. Calcium is sequestered in intracellular stores and rapidly captured and pumped back out of the cells subsequent to spiking. SNc neurons utilize L-type Ca2+ channels that assist in their pacemaking. These channels permit the entry of large amounts of calcium, but the SNc neurons have poor calcium buffering and handling capabilities. Thus, they are more susceptible to calcium overload and calcium-induced excitotoxicity than most other neurons.
9.3 Dopamine Modulates Striatal Synaptic Transmission
The circuitry responsible for Parkinson’s disease is centered in the striatum, which functions as the gateway to the basal ganglia. Striatal medium spiny projection neurons (MSNs) receive glutamatergic inputs from neurons in the cortex and thalamus. These centrally positioned neurons, in turn, project to a variety of cortical targets involved in body movement. Additional inputs are supplied to the MSN dendritic spines, and to the glutamate-releasing axon terminals providing the corticostriatal inputs, by dopamine-releasing axons from neurons located in the substantia nigra pars compacta (SNc). The loss of the dopamine releasing SNc neurons results in dysregulated synaptic transmission at the corticostriatal synapses. In particular, the motor control neurons receive malformed and excessively stimulatory signals from the striatal projection neurons that result in movement deficits and Parkinson’s disease.
In more detail, dopamine is a neuromodulator—molecules that produce changes in the electrical excitability of neurons and neural circuits. These molecules (serotonin and histamine are other examples of neuromodulators) bind members of the seven-pass, G-protein-coupled receptor (GPCR) family such as the metabotropic glutamate receptors (mGluRs) introduced in the previous chapter. Ligand binding to these GPCRs initiates a series of signaling events that regulate neurotransmitter detection by the NMDA and other ligand-gated ion channels on the postsynaptic side and neurotransmitter release properties on the presynaptic side. Calcium, sodium and potassium fluxes through the ion channels are altered and membrane electrical excitability is updated and adjusted according to the additional needs (Fig. 9.2). By these methods, dopaminergic inputs acting at presynaptic and postsynaptic sites regulate glutamatergic synaptic transmission.
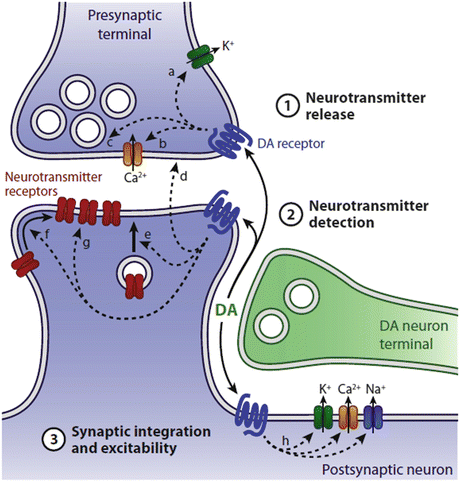
Fig. 9.2
Dopaminergic modulation of corticostriatal synaptic transmission. Release of dopamine from the SNc axon terminal binds dopaminergic, seven-pass G-protein-coupled receptors on pre- and postsynaptic terminals. These actions modulate neurotransmitter release from the presynaptic corticothalamic axon terminal and neurotransmitter detection and synaptic integration at the postsynaptic, striatal dendritic terminal (from Tritsch Neuron 76: 33 © 2012 Reprinted by permission from Elsevier)
9.4 Mutations and Overexpression of α-Synuclein Cause Autosomal Dominant Parkinson’s Disease
Alpha-synuclein has been discovered several times. It was first discovered in the neurons of the Torpedo californica electric ray by Maroteaux in 1988. Using an antiserum against cholinergic synaptic vesicles, the protein was identified in presynaptic terminals of neurons and portions of the nuclear envelop, hence its name synuclein. It was next uncovered in 1993 by Uéda as the precursor protein NACP, the NAC being a 35-amino acid fragment found to co-localize with Aβ in Alzheimer’s disease plaques but not derived from Aβ or its precursor. Hence it bears the appellation “non-Aβ component of the AD amyloid (or NAC).” Those findings were followed by the Spillantini and Polymeropoulos discoveries.
α-Synuclein is a vertebrate-specific protein highly expressed in the brain, especially in the neocortex, hippocampus and substantia nigra. Nowadays, it sits on top of the pantheon of high agents of Parkinson’s disease listed in Table 9.2. Parkinson’s disease is not the only neurodegenerative disorder characterized by α-synuclein-positive filamentous inclusions in neurons or glia. Rather, several neurological disorders, each with its own distinct set of clinical and pathological features, have α-synuclein-positive inclusions. The most prominent of the alpha-synucleinopathies are:
Dementia with Lewy bodies (DLB): Other names for this disorder are Lewy body dementia (LBD) and LB variant of AD (LBV/AD). Prominent among the clinical features of this disorder are extrapyramidal signs such as bradykinesia (slowness of movement), cognitive decline, visual hallucinations or delusions, and sensitivity to antipsychotic (neuroleptic) medications. Described originally by Okazaki in 1961 this has emerged as one of the most common forms of dementia although it is less prevalent than Alzheimer’s.
Multiple system atrophy (MSA): clinical features include parkinsonism along with autonomic and gait abnormalities, cerebellar signs, and cognitive decline. A major pathological feature is the presence of tubulofilamentous inclusion in oligodendrocytes referred to as glial cytoplasmic inclusions (GCIs). These are found in multiple regions of the brain.
Neurodegeneration with brain iron accumulation, type I (NBIA): This disorder is characterized clinically by parkinsonism accompanied by cognitive decline, cerebellar signs and bulbar symptoms. Its chief pathological feature is the buildup of iron and axonal swellings termed spheroids. GCIs and LBs are commonly encountered, as well.
9.5 Structure and Function of α-Synuclein
α-Synuclein possesses three domains. It has a positively charged N-terminal amphipathic region in which are embedded a series of KTKEGV repeats that mediate binding to negatively charged membranes. It possesses a central non-amyloid component (Uéda’s NAC) domain containing a stretch of hydrophobic residues responsible for its tendency to aggregate and form β-sheets, and it has a C-terminal acidic region. These domains and the locations of the six known PD-causing mutations are presented in Fig. 9.3.
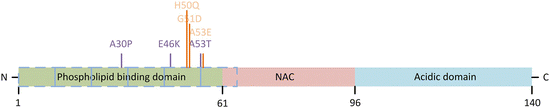
Fig. 9.3
α-Synuclein domain structure and disease-causing mutations. Shown are the N-terminal phospholipid/lipid raft binding domain, the central hydrophobic and aggregation-prone NAC, and the C-terminal negatively charged acidic domain. A series of repeats (depicted by the blue dashed boxes) in the N-terminal each contains an imperfect, KTKEGV-like consensus motif. The original three disease-causing mutations are depicted in purple and the recently found trio in orange
One of its most important properties is its ability to develop amphipathic α-helical secondary structure in its N-terminal region upon association with (negatively charged) lipid raft s . When doing so, the C-terminal region remains free and unstructured. Not only does α-synuclein associate with these membrane microdomains but also operates as a sensor of membrane curvature. This second key property enables the protein to preferentially associate with the more highly curved vesicles that package neurotransmitters in presynaptic terminals and other curved membranes such as those that bound mitochondria.
Like other intrinsically disordered protein s (discussed in Section 4.10) it is highly flexible and sensitive to environmental conditions. It has been called a “chameleon” for its ability to assume any of a number of alternative conformations depending on its immediate surroundings. α-Synuclein has been explored using a variety of techniques ranging from low resolution ensemble studies to high resolution single-molecule biophysics. In recent explorations using optical tweezer s , the monomeric protein (cytosolic form) was found fluctuate between large numbers of marginally stable structures, and possessed an energy landscape that closely resembled the schematic depicted presented by Uversky in Fig. 4.12b. That is, its energy landscape was flat and rugged with energy barriers on the order of 2–3 k B T. Those topographical features were in contrast to the funnel-shaped and smooth landscapes typical of well-folded globular proteins (Fig. 4.12a).
A new generation of optical microscopic methods have been developed that provide nanoscale images of living cells. These methods fill in the gap between low resolution live imaging and high resolution electron microscopy in nonliving tissue samples. The diffraction (λ/2) resolution limit of lens-based optical microscopes was put forth by the German physicist Ernst Abbe (1840–1905) in 1873. This limit has held sway until recently when new fluorescence method s such as stimulated emission depletion (STED) microscopy and fluorescence recovery after photobleaching (FRAP) were developed. These new developments overcome the diffraction barrier and make possible nanoscale imaging in living systems.
One of the areas being explored with these new probes is synaptic vesicle dynamics. Synaptic vesicles are approximately 40 nm in size and these reside in synaptic boutons on the order of 1 μm in diameter. The new methods reveal that while some vesicles are localized to a single synaptic bouton others form a pool that circulates between multiple boutons. By this means synapses can significantly alter their vesicle composition in a matter of minutes. Interestingly, it turns out that α-synuclein exerts considerable influence over vesicle dynamics and interacts with a variety of presynaptic vesicle-associated proteins. A number of these critical interactions had been identified leading to the emergence of a consistent picture of impaired vesicle dynamics and synaptic dysfunction arising from overexpressed (by a factor of 2 or 3) and mutated α-synuclein.
That α-synuclein interacts with proteins involved in synaptic vesicle formation and dynamics is supported by multiple studies. Neurotransmitter release occurs when neurotransmitter-laden synaptic vesicles fuse with the plasma membrane. This step is regulated by soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) and Sec1/Munc18-like proteins. Alpha-synuclein has been shown to promote assembly and disassembly of these essential complexes. Another key protein is the presynaptic vesicle chaperone, cysteine string protein-α (CSPα), which assists in the localization and exocytosis secretory vesicles containing neurotransmitters and peptides such as dopamine. CSPα helps direct chaperone complexes to the SNARE machinery and helps preserve the correct folding of proteins. α-Synuclein operates not only in conjunction with SNARE complexes but also cooperatively with CSPα. A third locus of α-synuclein activity is in its ability to inhibit vesicle trafficking from the endoplasmic reticulum to the Golgi. One of the earliest signs of Parkinson’s disease is the accumulation of clusters of vesicles indicative of emerging dysregulation and malfunctions in vesicle trafficking and dopamine handling. Sitting at the center of a cascade of events excessive alpha-synuclein helps to bring this about.
When mutated or overexpressed the protective activities of α-synuclein are lost and in their place the protein misfolds, aggregates, and promotes neurodegeneration. One of the main goals in the field has been and remains to identify the precise toxic forms of the molecule so that effective pharmacological treatments can be devised. According to the widely held toxic oligomer hypothesis the filamentous deposits found in the Lewy bodies are not the primary causal agents. Instead, the smaller oligomeric species are responsible. This hypothesis is supported by an impressive body of evidence. First, there is the poor correlation between the presence of Lewy bodies and Parkinson’s disease. Lewy bodies are commonly found at autopsy in the brains of elderly but otherwise neurologically normal brains. Secondly, oligomers generated through laboratory-induced genetic manipulations of SNCA reproduce many of the PD-associated presynaptic deficits. Thirdly, progress has been made in uncovering the biophysics of the transformations. The E46K and A53T mutations, in particular, produce deficits in presynaptic protection; they encode proteins that expose an increased fraction of hydrophobic surface area, and possess considerable β-sheet structure.
Mutated and overexpressed alpha-synuclein aggregates into a variety of oligomeric forms ranging in size from dimers, trimers and tetramers up to 100-mers or larger. These can assume a wide range of morphologies from linear and spherical structures to annular rings to large fibrillar assemblages. The latter, generated through nucleated-polymerization, can subsequently fragment and generate more oligomers. Each oligomeric species exists as an ensemble of conformations residing in a dynamic equilibrium and interconverting into one another, and into other oligomeric forms. Some of these conformers have a higher beta-sheet content and hydrophobicity than the others. Significantly, low and high β-sheet classes of conformers are separated by energy barriers that slow down the transition from harmless conformations to the toxic ones and may provide a pharmacological point of entry.
Excess, misfolded, damaged, aggregated and otherwise unwanted proteins are degraded by the ubiquitin-proteasome system, by macroautophagy , and in the case of selected (long-lived) proteins bearing a KFERQ-like motif by chaperone-mediated autophagy (CMA). The latter is a form of autophagy in which substrates are not packaged in vesicles, but instead, are sent one-by-one directly to and into lysosomes for degradation. These systems complement one another. For example, if a protein is too large to be handled by the proteasome it can be degraded by one of the autophagic pathways. If the protein has formed an aggregate it can still be encapsulated in an autophagosome and shipped for lysosomal degradation via macroautophagic transport. If there are circumstances where when one pathway becomes blocked, the other pathways can compensate for the loss by increasing their participation.
In a healthy cell, α-synuclein can be cleared both by macroautophagy and CMA. However, these systems fail in PD leading to a buildup of toxic oligomer s , large aggregates, and fibrils. While there is information for how PD forms of α-synuclein can impair macroautophagy through its disruption of the autophagosome assembly process, it is the breakdown of CMA at the lysosome that has elicited the most interest. That is because not only α-synuclein but also several other parkinsonism -associated mutated proteins negatively impact autophagy and lysosomal function. One of these is LRRK2. That protein and its blocking effect on CMA are examined next.
9.6 Mutations in LRRK2 Cause Autosomal Dominant Parkinson’s Disease
In 2002, Funayama identified a new locus for an autosomal dominant form of Parkinson’s disease. That locus carrying the PARK8 designation mapped to chromosome 12p11.2-q13.1. Two years later, in 2004, independent studies by Zinprich and Paisán-Ruíz identified the encoded protein, LRRK2, and several of its disease-causing mutations. Since many of the families examined by Paisán-Ruíz were of Basque descent, he named the protein “dardarin,” from the Basque word dardara, meaning trembling. A series of papers appearing shortly thereafter identified the G2019S mutation in LRRK2 as the single most commonly encountered mutation in PD sufferers. In some ethnic population groups, 30–40 % of all PD cases can be traced to that one mutation.
LRRK2 and alpha-synuclein both generate autosomal dominant forms of PD with similar features. The proteins, however, are strikingly different. While alpha-synuclein is small and contains large unstructured stretches, LRRK2 is huge and highly structured. The LRRK2 gene contains 51 exons, and encodes a protein containing 2527 amino acid residues organized into six well-defined domains (Fig. 9.4). This protein belongs to the so-called ROCO family of proteins containing not one but two distinct enzymatic activities—a guanosine triphosphatase (GTPase) activity (Ras-of-complex, Roc) and a kinase activity with a C-terminal of Ras (COR) domain in between. In addition to its enzymatic activities, the armadillo, ankyrin, leucine-rich repeat and WD40 domains confer on LRRK2 the ability to carry out protein–protein interactions and serve as a signaling scaffold.
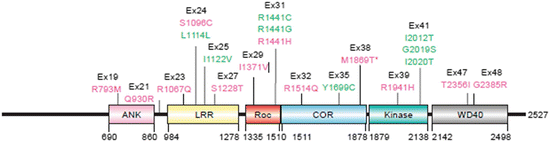
Fig. 9.4
LRRK2 domain structure and disease-causing mutations. Shown are the ankyrin (ANK), leucine-rich repeat (LRR), Ras-of-complex (Roc), C-terminal of Ras (COR), kinase, and WD40 domains. Not depicted: an armadillo domain N-terminal to the ankyrin domain. Disease-causing amino acid substitutions are denoted in green; other putative pathogenic sequences in magenta, and the corresponding exon numbers are indicated in black (from Mata Trends Neurosci. 29: 286 © 2006 Reprinted by permission from Elsevier)
The presence of two enzymatic activities is highly unusual and there is considerable controversy regarding their relationships and functioning. The most widely accepted model is one where the GTPase activates the kinase and the latter functions as the output unit. In that model, the GTPase must be in its ON state for the kinase domain to be catalytically active. Another requirement in this model is autophosphorylation. The functional form of LRRK2 is that of a dimer (oriented head-to-head), and this arrangement facilitates autophosphorylation at a number of sites including some located in the catalytically active site of the kinase. Most of the key disease-causing mutations are located in the central part of the protein. The two mutations occurring at sites in the Roc GTPase domain and the one located in the COR inhibit the GTPase but do not prevent the downstream kinase activity in a clear-cut manner, while the G2019S mutation located in the kinase domain actually enhances the kinase activity by a factor of three.
A further complexity arises from the presence of the protein–protein interaction domains. Their presence implies that LRRK2 functions as part of a signaling network with both upstream and downstream signaling partners. A number of these have now been identified among which are members of the 14-3-3 family of signaling modules. The importance of this interaction is derived from 14-3-3 ability to influence both the cellular location of LRRK2 and its propensity to aggregate. It turns out that phosphorylation at S910 and S939 sites located in between the ankyrin and LRR domains of LRRK2 is a crucial event that enables 14-3-3 binding. While either the kinase domain of LRRK2 or an upstream kinase can, in principle, carry out this task recent evidence points to one or more external kinases as the primary agents. Mutations such as R1441C/G/H and I2020T that abolish phosphorylation and 14-3-3 binding promote LRRK2 aggregation.
One major clue as to the functions of LRRK2 was the finding that the protein localizes to lipid raft s and caveolae . Recall that these specialized microdomains can be found embedded in the plasma membrane and intracellularly in organelle and vesicle membranes. LRRK2 can be detected in all of these locales. Recent studies emphasized possible locations in autophagic vacuoles (AVs) and in intraluminal vesicles (ILVs) of multivesicular bodies (MVBs ), while earlier studies hinted at localization in lysosomes, endosomes, the mitochondrial outer membrane, the Golgi, the plasma membrane, and synaptic vesicles. At these sites, LRRK2 participates in the biogenesis and/or regulation of the intracellular membranous structures. Of special note is its role in autophagy; it has been found to regulate autophagic vacuole activity, clearance of trans-Golgi-derived vesicles through the autophagic-lysosomal pathway, and CMA (to be discussed in the next section). In addition, it plays a role in presynaptic vesicle storage and mobilization.
9.7 Chaperone-Mediated Autophagy Is Blocked by Mutated α-Synuclein and LRRK2
Recall from Chap. 6 that in autophagy unwanted proteins, protein aggregates, and organelles are sent to lysosomes for hydrolytic degradation. CMA differs from macroautophagy (autophagy) and microautophagy in two ways. First, rather than packaging the substrates in transport vesicles the proteins targeted for degradation in CMA are sent one-by-one directly through the lysosomal membrane and into the hydrolytic compartment for degradation. Secondly, in autophagy and microautophagy the targeting of misfolded and damaged proteins is nonspecific, while CMA targets only a specific subset of proteins. The main elements of CMA were uncovered in the late 1980s and early to mid1990s and motivated by a suggestion by J. Fred Dice that there might be a form of autophagy that targeted specific proteins for degradation in lysosomes. Experiments carried out in that time period in several groups and especially by Dice, Ana Maria Cuervo, and their coworkers led to the uncovering of the main elements of this pathway and confirmed Dice’s prescient surmise.
The main elements of CMA consist of a recognition sequence, a chaperone, and a receptor. Proteins destined for degradation by CMA contain a KFERQ-like sequence. Motifs of this type are found on about 30 % of all the proteins. These sequences are recognized by heat shock cognate protein of 70 kDa (Hsc70), a constitutively expressed member of the Hsp70 family of molecular chaperones. The chaperone-bound substrates are then conveyed to the lysosome where they attach to the LAMP-2A receptors. As is the situation discussed in earlier chapters, proteins are translocation across the membrane in an unfolded form. In these operations, depicted in Fig. 9.5, newly synthesized LAMP-2A receptors reside in lipid raft s where (1) their mobility is restricted and where after a short time they are degraded, or (2) CMA is activated and the LAMP-2A receptors exit the rafts to carry out their translocation tasks.
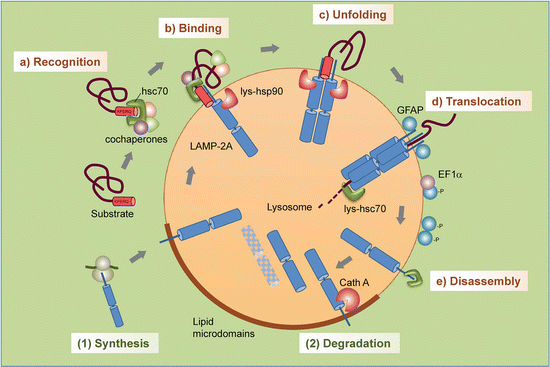
Fig. 9.5
Steps in chaperone-mediated autophagy : (a) Recognition of substrate proteins by hsc70/cochaperones; (b) binding of substrate-chaperone complex to LAMP-2A; (c) unfolding of the substrate; (d) LAMP-2A multimerization and substrate translocation followed by lysosomal degradation; (e) disassembly of LAMP-2A complex. Regulation of LAMP-2A at the lysosomal membrane occurs through (1) de novo synthesis and (2) sequestration and degradation in lipid raft s (from Kaushik Trends Cell Biol. 22: 407 © 2012 Reprinted by permission from Elsevier)
One of the ways that PD-associated α-synuclein and LRRK2 exerts their toxic effects is through their jamming of the LAMP-2A-apparatus. Not only do these blocking actions impair the removal of other damaged monomers and aggregates of the same species, and of each other, so they build up to toxic levels, but they also impair removal of other damaged and unwanted proteins. A particularly revealing example of the negative effects of blocking CMA by mutated α-synuclein or LRRK2 is provided by the blocking of neuronal survival factor MEF2D (myocyte enhancer factor 2D). This transcription factor is required for neuronal survival. It continuously shuttles to the cytoplasm where it binds to Hsc70 and is then transported to lysosomes for degradation. Under normal conditions CMA removes inactive MEF2D from the active pool and by that means contributes to survival. The blocking by α-synuclein/LRRK2 prevents this clearing operation.
9.8 The PINK1/Parkin Pathway Regulates Mitophagy
Badly damaged and unrepairable mitochondria undergo mitophagy , a specialized form of macroautophagy in which mitochondria are encapsulated in autophagosomes and shipped to lysosomes for degradation. That disposal route is part of a mitochondrial quality control system that includes mitochondrial chaperones that refold misfolded proteins, and proteases that degrade unrecoverable ones. It includes the machinery to ship outer membrane proteins to the ubiquitin-proteasome system for disposal, and the machinery to initiate mitophagy that removes the remainder.
In order for mitophagy to take place, a damaged mitochondrion must be isolated from the remaining healthy mitochondrial network. That step involves activating the mitochondrial fission machinery and inactivating the fusion apparatus. The protein Drp1 is a critical mediator of fission and has to be activated. It organizes a multiprotein complex that encircles the outer mitochondrial membrane and exerts mechanical scission forces. Three GTPases—OPA1 functioning at the inner membrane and Mfn1 and Mfn2 at the outer membrane—regulate fusion. These need to be inactivated and the inner membrane protease OMA1 activated.
Mutations in the PARK2 that encodes parkin and the PARK6 gene that encodes PINK1 head up the list of risk factor s for developing autosomal recessive forms of Parkinson’s disease (ARPD). These proteins are essential components of the mitochondrial quality control system. That parkin was a cause of ARPD was discovered by Kitano in 1998. The discovery that PINK1 was also a cause of ARPD followed a few years later by Valente in 2004. A landmark in understanding the causes of ARPD was the realization shortly thereafter that both parkin and PINK1 operate in a single pathway. In that pathway, PINK1 functions as a sensor of mitochondrial damage and parkin as its effector, facilitating removal of oxidized and damaged proteins, or alternatively, mediating removal of the damaged organelle.
This pathway operates in the following way. PINK1 is a 581-amino-acid-residue serine/threonine kinase. It possesses an N-terminal mitochondrial targeting sequence and a serine/threonine kinase domain. This protein, like other proteins bearing a mitochondrial targeting sequence, is transported into mitochondria by transporter of the outer membrane (TOM) and transporter of the inner membrane (TIM) complexes. Under normal conditions, PINK1 is acted upon by one or more mitochondrial proteases such as presenilin-associated rhomboid-like protease (PARL) or mitochondrial processing peptidase (MPP) and degraded. Under abnormal membrane potential (ΔΨ m) conditions PINK1 is not degraded but instead accumulates and is stabilized on the outer mitochondrial membrane with its kinase domain facing the cytoplasm primed to recruit parkin. This situation is depicted in Fig. 9.6.
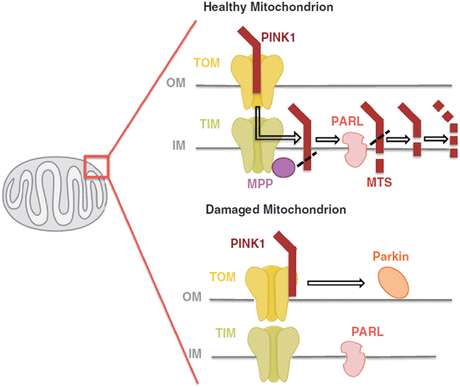
Fig. 9.6
PINK1 is imported into the inner mitochondrial membrane via the TOM/TIM complexes. The TIM complex-associated protease, mitochondrial MPP, cleaves the PINK1 mitochondrial targeting sequence (MTS). PINK1 is also chopped into pieces by the inner membrane presenilin-associated rhomboid-like (PARL) protease and proteolytically degraded. Loss of membrane potential prevents the import of PINK1 leading to the accumulation of unprocessed PINK1 on the outer membrane surface where it associates with the TOM complex and recruits cytosolic parkin to the damaged mitochondria (from Ashrafi Cell Death Diff. 20: 31 © 2013 Reprinted by permission from Macmillan Publishers Ltd)
Parkin is a 465-amino-acid-residue E3 ubiquitin ligase. It contains an ubiquitin-like Ubl domain in its N-terminal, and a series of motifs in its C-terminal that provides an E3 ubiquitin ligase capability to the protein. A long-held mystery was how PINK1 recruits parkin to the mitochondrial outer membrane. That mystery is being solved. Among the recent findings is that stabilized and activated PINK1 phosphorylates mitofusin 2 (Mfn2). This OMM protein is subsequently ubiquitinated by parkin and these operations are consistent with Mfn2 participating in the recruitment and serving as a parkin receptor. That finding was followed in early 2014 by reports that ubiquitin is phosphorylated by PINK1. This action may well be a central one as it not only appears to facilitate the recruitment of parkin to the mitochondrial outer membrane but also activates the ubiquitin ligase. That finding followed earlier studies in which PINK1 was found to phosphorylate parkin at the corresponding site, Ser65, in its amino-terminal Ubl domain. That step primes the E3 ligase for subsequent activation by phospho-ubiquitin.
Once parkin relocates to the mitochondrial outer membrane from the cytosol and is activated it ubiquitinates its substrates. Progress has been made on identifying parkin’s targets and understanding how their ubiquitination triggers mitophagy and other neuroprotective actions. Consistent with the needed fission/fusion steps, parkin ubiquitinates OPA1 and Drp1, and both PINK1 and parkin tag a protein called Miro for degradation. That protein tethers the OMM to kinesin motor protein s and its degradation arrests the movement of the mitochondrion. These are first steps in removing damaged mitochondria from the remaining healthy parts of the mitochondrial network, and begin the process of taking apart and removing the damaged mitochondria. The outer membrane proteins are then shipped to the 26S proteasome for degradation. That step is followed by mitophagy of the inner membrane and matrix proteins.
9.9 Neuroprotective Actions Take Place Independent of Mitophagy
PINK1 and especially parkin serve in a broad neuroprotective capacity. While many of their protective activities involve mitophagy , others occur independently of autophagy. These additional activities are launched in instances where they damage to the mitochondria is modest and not irrevocable. Some of these additional protective steps involve the regulation of gene expression.
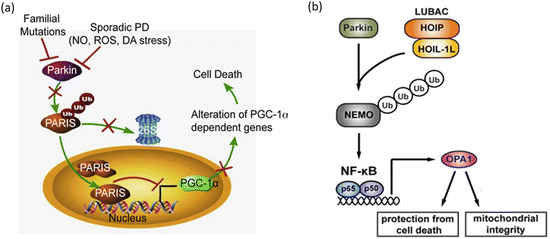
One of the first parkin substrates to be identified was a protein named PARIS. That protein accumulates in the brains of PD patients suffering from parkin-inactivating mutations, and also in sporadic PD when parkin is inactivated through oxidative stress es (Fig. 9.7a). PARIS acts as a transcription repressor and its primary substrate is PGC-1α, a master regulator of mitochondrial function and ROS responses. Repression of PGC-1α by PARIS prevents these actions and dopaminergic neurons die off. Another of parkin’s substrates is NEMO, a regulatory subunit of the NF-κB’s upstream activating complex. NF-κB is a central stress-activated transcription factor. One of the proteins whose transcription is increased by parkin-mediated ubiquitination of NEMO is the mitochondrial fusion regulator OPA1. That action protects the mitochondria from apoptotic cell death as depicted in Fig. 9.7b.