Fig. 6.1
(a) H&E staining of a substantia nigra neuron containing a Lewy body; (b) The core of each Lewy body stains more strongly for α(alpha)-synuclein than the characteristic halo (c) which is strongly immunoreactive for ubiquitin
Staining for Lewy pathology with antibodies to α(alpha)-synuclein indicates that the first location of pathologic change is in the olfactory apparatus and caudal brainstem, especially the dorsal motor nucleus of the vagus in the medulla. Neural involvement is thought to spread progressively rostrally up the brainstem in a fashion hypothesised by Braak and colleagues, who studied the pattern of α(alpha)-synuclein involvement in autopsied brains. The cerebral cortex is involved late in this schema, in keeping with the evolution of cognitive impairment, (if not frank dementia) in patients with long-standing IPD. When the motor symptoms of IPD are evident, the substantia nigra already has lost about 60 % of dopaminergic neurons, and the dopamine content in the striatum is about 80 % less than normal. Involvement of non-dopaminergic neurons including cholinergic neurons in the nucleus basalis of Meynert, noradrenergic neurons in the locus coeruleus and serotonergic neurons in the midline raphe may be significant in the non-motor symptoms.
6.2 Diagnosis
The diagnosis of IPD remains essentially a clinical one. If made by a neurologist, the diagnosis based on clinical impression has been shown to have a positive predictive value of 76 % up to 98.6 % for those working in a specialist movement disorders service. The United Kingdom Parkinson’s Disease Society Brain Bank criteria are typically used in Research studies of PD; bradykinesia with one of tremor, rigidity and postural instability are required in the absence of exclusion criteria (Table 6.1). Retrospective application of these criteria to patients diagnosed with PD in life demonstrates positive predictive values of between 82 and 92 %. This diagnostic accuracy may be improved if a levodopa response and asymmetry are also sought but sensitivity may be lost.
Table 6.1
Exclusion criteria for Parkinson’s disease
History of repeated strokes with stepwise progression |
History of repeated head injury |
History of definite encephalitis |
Oculogyric crisis (unless drug induced) |
Neuroleptic exposure at time of diagnosis |
Sustained remission |
Supranuclear gaze palsy |
Cerebellar signs |
Early severe autonomic involvement |
Early severe dementia |
Babinski sign |
Presence of cerebral tumour or communicating hydrocephalus on imaging |
Failure to respond to an adequate dose of levodopa (up to 2000 mg) |
Scans without evidence of dopaminergic deficit (SWEDDs) |
Some physicians will use the ‘levodopa challenge’ where a response to a single dose of up to 300 mg of levodopa supports the diagnosis of PD. Tremor predominant forms of IPD may not however demonstrate any response to levodopa and some atypical forms of parkinsonism will, thus causing diagnostic confusion. Others avoid this challenge, particularly in younger patients, given concerns that even a single dose of levodopa may ‘prime’ the basal ganglia for dyskinesia.
An important aspect of the initial and subsequent clinical assessments is to look for atypical features suggesting an alternative diagnosis, having important implications for predicting survival and treatment response. Some conditions mimicking IPD will require alternative treatment strategies (Table 6.2). Also, it is not uncommon for IPD to present with symptoms not readily attributed to the disease. Some of these patients will carry alternative diagnoses before the more obvious parkinsonian features appear (Table 6.3).
Table 6.2
Clinical features of the Parkinson-plus (atypical) disorders
Progressive supranuclear palsy |
Early falls |
Prominent axial rigidity |
Pure freezing of gait and early freezing |
Arm abduction when walking |
Frontalis overactivity (startled appearance) |
Deep naso-labial folds |
Vertical gaze palsy or ‘round the houses’ vertical saccades |
Blepharospasm |
Prominent Square-wave jerks |
Slowing of horizontal saccades |
Apraxia of eye opening |
Characteristic voice is a hoarse, throaty growl, with some hesitation between words |
Multiple systems atrophy |
Prominent cerebellar or autonomic features |
Flexed posture |
Anterocollis |
Myoclonus or polyminimyoclonus |
Laryngeal stridor (may only be nocturnal) |
Early orofacial dyskinesia with levodopa |
Pyramidal tract signs (e.g. extensor plantar responses, spastic ‘catch’ in addition to rigidity) |
Purple discolouration of the feet due to abnormal vascular autonomics |
Corticobasal degeneration |
Unilateral dystonia |
Alien-limb phenomenon |
Unilateral stimulus sensitive myoclonus |
Cortical sensory loss |
Dyspraxia |
Table 6.3
Parkinsonian symptoms and signs commonly attributed to other disorders
Fatigue |
Dyspnea |
Bradyphrenia |
Depression |
Joint pain (particularly shoulder pain) |
‘Radicular’ pain (true radicular pain may worsen in ‘off’ states) |
Foot cramps/dystonia |
Dysphonia |
Anxiety/panic attacks |
‘Weakness’ affecting ability to rise from chairs or apparently unilateral weakness |
6.3 Subtypes of Parkinson’s Disease
Parkinson’s disease can be classified into subtypes. The most common classification is based on the time of onset of PD: young versus late onset and secondly on the dominant feature: tremor-predominant versus akinetic-rigid phenotype with the mixed category falling in between those two. Below are discussed the main features of the different subtypes.
1.
Young onset Parkinson’s disease: age of onset between 20 and 40, with associated rigidity and dystonia, good response to L-dopa, but with an early development of dyskinesias, slower progression of the disease than in the late-onset PD. Associated with Parkin mutation (discussed in a genetics section) in 1/3 of the cases.
2.
Late-onset Parkinson’s disease: age of onset over 60, with a rapid disease progression.
3.
Tremor-predominant PD: often misdiagnosed as an essential tremor, good prognosis, with a slow progression.
4.
Postural instability and gait difficulty (PIGD) also called akinetic-rigid subtype: with an early cognitive decline and higher frequency of dementia, depression and apathy.
5.
Mixed PD: features of tremor predominant and PIGD.
6.4 Differential Diagnosis of Parkinsonism
6.4.1 Atypical Parkinsonism
Approximately three quarters of patients presenting with parkinsonism have typical motor features, and are most likely to have pathologically confirmed IPD. The remaining 25 % of patients will have so-called atypical parkinsonism, also called Parkinson-plus syndromes. This group of primary degenerative parkinsonian disorders includes progressive supranuclear palsy (PSP), multiple system atrophy (MSA) and corticobasal degeneration (CBD) and are covered in Chap. 9. All may start with an asymmetrical clinical syndrome indistinguishable from IPD. These forms of parkinsonism all share a tendency to be poorly responsive to levodopa, be largely symmetrical (with the exception of corticobasal degeneration) and have little or no rest tremor (although myoclonus mimicking tremor may be evident). Table 6.2 highlights clinical features that should raise suspicion of an atypical parkinsonism.
6.4.2 Dementia with Lewy Bodies
Patients presenting with dementia before, or within 1 year of manifesting parkinsonism, are by convention given a diagnosis of dementia with Lewy bodies (DLB). Visual hallucinations are common and the course of cognitive impairment is typically fluctuating, often with dramatic variability from 1 day to the next. Some patients will have prominent autonomic dysfunction. Patients with dementia beginning after 1 year are diagnosed with PD with dementia (PDD). Both these conditions may represent different points on the spectrum of ‘Lewy body disease’ with a larger cortical burden of Lewy bodies than in patients with IPD.
6.4.3 Secondary Parkinsonism
6.4.3.1 Drug-Induced Parkinsonism
Parkinsonism can follow exposure to drugs with an antagonistic effect at D2 receptors. This is the most common cause of secondary parkinsonism and is typically seen in patients requiring antipsychotic (neuroleptics, major tranquillizers) treatment. Newer, ‘atypical’ neuroleptics with less affinity to the D2 receptor are less likely to result in extrapyramidal side-effects and are preferred when treating psychosis in IPD. The commonly used anti-emetic drugs metaclopramide and prochlorperazine also have a D2 antagonist effect. Other drugs known to induce parkinsonism include lithium, tetrabenazine, reserpine, valproate, and the calcium channel blockers, cinnarizine and flunarizine.
Drug withdrawal typically results in a slow improvement although latent parkinsonism may have been unmasked and full recovery may not occur.
6.4.3.2 Vascular Parkinsonism
This is also known as ‘lower body parkinsonism’ due to prominent gait disturbance and relatively less arm involvement. Often, these patients will have early freezing which is not typically seen in IPD. This is an important cause of parkinsonism in older patients and those with a history of vascular risk factors (particularly hypertension). The pathophysiology is related to small vessel disease with prominent periventricular ischemia. Patients with basal ganglia infarcts are more likely to respond to levodopa. Magnetic resonance imaging (MRI) of brain is useful to identify those patients who may have a vascular cause of parkinsonism. Other clinical features that can help differentiate vascular from idiopathic parkinsonism are a postural more than resting tremor and preserved olfaction.
6.4.3.3 Fragile X Pre-mutation
The pre-mutation state of Fragile X can present with tremor, parkinsonism and autonomic features and may therefore be misdiagnosed as essential tremor, IPD or MSA. An accurate family history is vital, looking for a history of a related child with learning disability or autism. The presence of ataxia is another important clue. In one series of 26 patients with premutations of the FMR1 gene, 57 % of cases had mild bradykinesia, resting tremor was present in 40 % and 71 % had upper limb rigidity.
6.4.3.4 Others
Secondary parkinsonism can also occur following toxin exposure, including manganese (miners, intravenous drug abuse), carbon disulphide and 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (described by Langston JW and Palfreman J in “The Case of the Frozen Addicts”).
Rarely parkinsonism can arise as a consequence of strategically-placed structural lesions such as large Virchow-Robin (perivascular) spaces or central nervous system (CNS) tumors, more commonly supratentorial meningiomas causing basal ganglia compression than by direct tumor infiltration.
Functional parkinsonism is well recognized but rare. Clues to the diagnosis are a history of previous psychogenic illness, an abrupt onset, entrainment of tremor, selective disability and distractibility.
6.4.4 Disorders That Can Mimic Parkinsonism
6.4.4.1 Essential Tremor
Essential tremor (ET) is one of the most common disorders mistaken for IPD, characterized by a postural and kinetic tremor without rest tremor. Patients with ET can have cog-wheeling but without rigidity. Where there is a combination of a resting hand tremor with essential tremor, the physician should consider rest tremor appearing late in ET, or the combined resting-postural tremor syndrome.
Parkinsonism and essential tremor could also represent the co-occurrence of two common movement disorders.
6.4.4.2 Dystonic Tremor
Dystonic tremor usually occurs in a dystonic body part. Some distinguish this from ‘dystonia with tremor’, tremor observed in an unaffected body part with dystonia elsewhere. Dystonic upper limb tremor will sometimes have a ‘null-point’ where rotation of the affected limb will reach a point where the tremor is abolished. Like ET, dystonic tremor of the upper limbs will not have the latent period before re-emerging on changing position as seen in IPD (re-emergent tremor). Dystonic tremor tends to be more irregular and jerky in character and may have a torsional component.
6.4.4.3 Normal Pressure Hydrocephalus
Normal pressure hydrocephalus (NPH) presents with one or all features of a triad of gait apraxia, urinary incontinence and cognitive impairment. Gait can be similar to that of vascular parkinsonism because of involvement of periventricular descending corticospinal tracts. Imaging is essential in demonstrating dilatation of all ventricles out of proportion to the degree of cortical atrophy. Diagnosis is made most reliably by removal of a large volume (at least 40 ml) of cerebral spinal fluid (CSF) via lumbar puncture, which can also predict the potential for improvement with shunt placement although this remains controversial. Video of gait and cognitive assessment performed pre and post lumbar puncture is useful for later assessment.
6.4.5 The Role of Imaging in Diagnosing Parkinson’s Disease
With a classical clinical picture, there is little or no role for neuroimaging in making a diagnosis of PD. Positron emission tomography (PET) with the fluordopa ligand and single photon emission computed tomography (SPECT) are the principal options.
In SPECT studies, radioligands of the dopamine transporter (DAT) are used to determine the pre-synaptic integrity of nigrostriatal neurons. The DAT is exclusively localised to dopamine-producing neurons. Advantages of the technique are the wide availability of SPECT scanners and the ability to continue dopaminergic medication at the time of imaging. Patients with IPD will demonstrate reduced radiotracer uptake in the striatum bilaterally which tends to be asymmetrical, particularly affecting the posterior (dorsal) putamen (Fig. 6.2). Scans without evidence of dopaminergic dysfunction (SWEDDs) is the term applied to normal scans of patients with a clinical diagnosis of IPD. The diagnosis in these patients likely represents a false positive as no long-term data or post-mortem studies have subsequently proven a diagnosis of IPD. Many of these patients will have a true diagnosis of essential or dystonic tremor, and some may have dopa-responsive dystonia, in which parkinsonism is often a feature, but the clue is the DAT scan is normal.
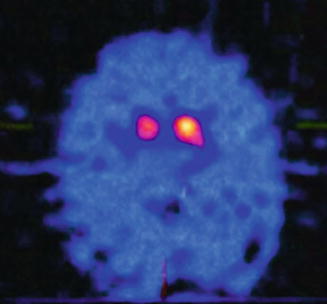
Fig. 6.2
Transaxial sections of a I-123 Ioflupane SPECT (DaTSCAN) from a patient with idiopathic Parkinson’s disease demonstrating bilateral loss of uptake in the posterolateral aspect of the putamen bilaterally in a pattern typically seen soon in early disease
SPECT imaging has no role in differentiating atypical parkinsonism from IPD, because both have reduced DAT imaging. Its main use is in differentiating IPD from ET, drug-induced tremor/parkinsonism or psychogenic tremor, all of which should have normal imaging. Transcranial sonography has emerged as an alternative imaging modality, with nigral hyperechogenicity having a sensitivity of up to 90 % for IPD. Correlation with disease stage or severity has not been proven, and the significance of abnormalities in approximately 10 % of clinically unaffected individuals has yet to be established.
While the DAT scan remains the only approved PD diagnostic tool, recent research shows the possibility of a 3 T-susceptibility-weighted (SWI) MRI being a new accurate test for PD. The healthy nigrosome-1 (largest of the five described) is easily visualized on 3 T SWI as a presence of a ‘swallow tail’ of the dorsolateral substantia nigra, which is absent in PD. Resting state fMRI also holds a promise to aid the early diagnosis of PD with the latest research showing reduced resting functional connectivity in the basal ganglia in PD patients with an intact cognition.
6.5 Genetics
Case–control studies have confirmed a higher prevalence of IPD amongst first-degree relatives of affected patients supporting a genetic component to the disease. However the relative contribution of environmental and genetic factors to the pathophysiology of idiopathic PD is unclear. A number of Mendelian single gene mutations are associated with familial clustering of Parkinson’s disease, although this accounts for less than 10 % of all PD.
Familial PD has both clinical and pathological overlap with IPD but commonly has a younger age at onset. The first single gene mutation identified 17 years ago as a cause of familial PD was in the gene coding for α(alpha)-synuclein. There has been more recent interest in the study of common variants or single nucleotide polymorphisms (SNPs) in the genes associated with familial PD. Common variants may be associated with an increased risk of sporadic PD although effect sizes are small and larger study populations are required to adequately power case–control studies. Some of the genes and their products associated with familial PD are discussed below.
6.5.1 α(alpha)-synuclein (PARK 1)
The SNCA gene encoding the α(alpha)-synuclein protein is located on chromosome 4q21.3. α(alpha)-Synuclein is an abundant presynaptic protein of unclear function. The resulting parkinsonism transmits in an autosomal dominant pattern. It is rare, being reported only in a handful of families from Greece, Italy, Germany, and Spain. The protein, α(alpha)-synuclein, is present in Lewy bodies. Duplication and triplication of the α(alpha)-synuclein gene also causes familial parkinsonism (PARK4), indicating that over-expression of the normal (wild-type) synuclein protein is sufficient to provoke dopaminergic neurodegeneration. This supports a pathogenic role for α(alpha)-synuclein in IPD. There is debate as to whether Lewy bodies are contributing to the pathogenesis of PD or if the aggregation of α(alpha)-synuclein fibrils to form Lewy bodies is an effort of the cell trying to protect itself from toxic α(alpha)-synuclein oligomers.
6.5.2 Parkin (PARK 2)
The Parkin gene is found on chromosome 6q25.2–27 and is the most common genetic cause for early-onset PD (before age 50), accounting for 50 % of familial and 20 % of sporadic early onset disease. Parkin mutations give rise to autosomal recessive PD that can have typical features of IPD, but may also demonstrate hyperreflexia, dystonia at presentation and sleep benefit. Rest tremor is not prominent. Post-mortem studies have shown nigral degeneration in patients with Parkin mutations without Lewy bodies. There is an ongoing debate whether the heterozygote carrier state predispose to later onset PD in families.
6.5.3 PINK1 (PARK 6)
After parkin mutations, PINK1 mutations are the second most common cause of early onset PD, sharing autosomal recessive inheritance. Disease progression is usually slow with early levodopa-induced dyskinesias. The parkinsonism is often preceded by anxiety and depression. The PINK1 gene codes for a mitochondrial protein that is a recognized component of Lewy bodies seen in late onset IPD, and the few available autopsy studies have identified typical neuropathological findings.
6.5.4 DJ-1 (PARK 7)
Mutations in the DJ–1 account for 1–2 % cases of early-onset familial PD. The presentation is with a typical early onset parkinsonism, often with dystonic and neuropsychiatric features. Unlike the unaffected heterozygous state with Parkin and PINK1 mutations, carriers do not demonstrate functional neuroimaging evidence of nigro-striatal dysfunction.
6.5.5 LRRK2 (PARK 8)
PARK8 is mapped to chromosome 12q12 and encodes for a previously unknown protein named leucine-rich repeat kinase-2 (LRRK2), ubiquitously expressed in the CNS. Seven pathogenic LRRK2 mutations have been found, and are the most frequent genetic cause of familial PD. They account for up to 5 % of sporadic PD in the Caucasian population. In Ashkenazi Jews and North African Berber Arabs, LRRK2 mutations have been found in up to 20–40 % of both familial and sporadic cases of PD. The most prominent mutation in the Caucasian population is the G2019S substitution. LRRK2 mutations result in an autosomal-dominant parkinsonism that resembles typical late-onset IPD. Cognitive impairment is usually not a feature. Although the neuropathology associated to LRRK2 mutations is highly variable, degeneration of substantia nigra neurons has been consistently observed.
6.5.6 Others
Glucocerebrosidase (GBA) gene mutations, when homozygous, cause autosomal recessive Gaucher’s disease. Heterozygous carriers are at increased risk of developing parkinsonism that is indistinguishable from IPD. Up to 30 % of Ashkenazi Jews with PD have been found to have this mutation; the mutation causes PD in other ethnic groups as well. Dopa-responsive dystonia may present during adulthood as slowly progressive parkinsonism and tends to responds to low doses of levodopa. Parkinsonism can also be a predominant feature of the Westphal variant of Huntington’s disease, although this is usually in juvenile patients and family history should be informative. Some forms of spinocerebellar ataxia (SCA2 and SCA3) can present with a levodopa-responsive parkinsonism with minimal cerebellar features.
Frontotemporal dementia linked to chromosome 17 (FTDP-17) can present with parkinsonism especially the pallido-ponto-nigral degeneration (PPND) variant, which is also associated with insomnia square wave jerks and a supranuclear gaze palsy.
6.6 Clinical Features
6.6.1 Motor
6.6.1.1 Rest Tremor
Rest tremor, typically of 4–5 Hz, is the first symptom recognized in 70 % of patients, but may be absent in 20 %. The classic “pill-rolling” tremor involves the thumb and forefinger and is best seen when the patient is walking. Rest tremor disappears with action but re-emerges after a latent period of seconds as the limbs maintain a posture (re–emergent tremor). Tremor increases with walking (a possible early sign), stress or excitement. Tremor is also common in the lips, chin, and tongue but not the head.
6.6.1.2 Bradykinesia with Decrement
Bradykinesia encompasses slowness of movement, difficulty initiating movement and loss of automatic movement. Decrement refers to a reduction in amplitude of movement, particularly with repetitive movements. Often different tactics need to be used by the examiner to bring out bradykinesia that might only be seen during certain actions, such as finger tapping, pronation-suppination movements or opening and closing the fists. The face loses spontaneous expression (hypomimia) with decreased frequency of blinking. Speech becomes soft (hypophonia), and the voice has a monotonous tone with a lack of inflection (aprosody). Some patients do not enunciate clearly (dysarthria) and do not separate syllables clearly, thus running the words together (tachyphemia) and others stutter (palilalia). Bradykinesia of the dominant hand results in small and slow handwriting (micrographia). Difficulty rising from a deep chair, getting out of cars and turning in bed are symptoms of truncal bradykinesia. Subtle signs of bradykinesia can be detected by examining for slowness in shrugging the shoulders, smiling, lack of natural gesturing in conversation and decreased blink frequency. Walking is slow, with a shortened stride length and a tendency to shuffle with decreased heel strike; arm swing decreases and eventually is lost.
6.6.1.3 Rigidity
Rigidity is an increase of muscle tone on passive movement and is not velocity dependent as seen with spasticity. Resistance is equal in all directions and usually has a ‘cogwheeling’ character caused by the underlying tremor even if not visible. Rigidity of the passive limb increases while another limb is engaged in voluntary active movement, also known as the co-activation or facilitation test. Axial rigidity at the neck can similarly be accentuated by asking the patient to open and close both hands. Mild upper limb rigidity can be elicited by standing behind the patient and rocking their shoulders back and forward to produce passive arm swing that will be reduced on the more affected side.
6.6.1.4 Loss of Postural Reflexes
Loss of postural reflexes leads to falling and eventually to an inability to stand unassisted. These reflexes are tested by the pull-test during which the examiner, who stands behind the patient, gives a sudden firm pull on the shoulders after explanation of the procedure, and checks for retropulsion. With advance warning, an unaffected person can recover within two steps.
6.6.1.5 Flexed Posture
This commonly begins in the elbows and spreads to involve the entire body. The head is bowed, the trunk is bent forward, the back is kyphotic and the arms are held in front of the body with the elbows, hips, and knees flexed. Walking is marked by festination, whereby the patient walks faster and faster with short steps, trying to move the feet forward to be under the flexed body’s center of gravity to prevent falling. Deformities of the hands include ulnar deviation, flexion of the metacarpophalangeal joints, and extension of the interphalangeal joints (striatal hand). The hallux may be dorsiflexed (striatal toe). Lateral tilting of the trunk can develop (Pisa syndrome) and extreme flexion of the trunk (camptocormia) is sometimes seen which should be abolished when lying flat.
6.6.1.6 Freezing
This manifests as the transient inability to perform active movements. Freezing occurs suddenly and is transient, usually lasting seconds. It will typically occur when the patient begins to walk (start hesitation), attempts to turn while walking, approaches a destination, such as a chair in which to sit (destination hesitation). Tight spaces can also provoke freezing, such as doorways, as can time-restricted activities such as crossing heavily trafficked streets or answering the phone. The combination of freezing and loss of postural reflexes is particularly devastating, and a common cause of falls.
6.7 Non-motor Symptoms
Later in the clinical course, non-motor and axial motor symptoms become prominent and account for greater disability, being poorly responsive to dopaminergic treatment (Table 6.4). After 20 years of disease in the Sydney Multicentre Study, falls were experienced by 87 %, moderate dysarthria in 81 %, dementia in 84 %, visual hallucinations in 74 %, postural hypotension in 48 % and urinary incontinence in 71 %. Some non-motor symptoms can be observed as ‘pre-motor’ phenomena, appearing before typical motor features. These include constipation, rapid-eye-movement (REM) sleep behavior disorder, olfactory impairment and mood disorders. Some of the more troublesome problems and their management are discussed below.
Table 6.4
Non-motor symptoms in Parkinson’s disease
Neuropsychiatric |
Depression |
Anxiety, panic attacks |
Hallucinations, illusions, delusions |
Dementia, mild cognitive impairment |
Obsessional, repetitive behaviorsa |
Deliriuma |
Anhedonia |
Autonomic symptoms |
Orthostatic hypotension |
Nocturia, urgency, frequency |
Paroxysmal sweating |
Seborrhea |
Erectile impotence |
Xerostomia |
Gastrointestinal |
Ageusia |
Sialorrhea |
Nausea and vomiting |
Dysphagia |
Constipation |
Incontinence |
Sensory symptoms |
Pain (can be pseudoradicular) |
Paraesthesia |
Olfactory disturbance |
Visual blurring |
Sleep disorders |
REM sleep behavior disorder |
Difficulty initiating or returning to sleep, insomnia |
Restless legs syndrome |
Periodic limb movements in sleep |
Vivid dreaming |
Nocturnal hallucinations |
Excessive daytime somnolence |
Others |
Fatigue |
Seborrhea |
Weight loss or gaina |
6.7.1 Autonomic Involvement
6.7.1.1 Constipation
Constipation is almost universal in PD and can influence the efficacy of oral therapies by causing erratic absorption. Treatment with a regular stool softener, sometimes combined with a stimulant laxative is usually effective and most patients will require a regular laxative. The use of abdominal plain films can guide the use of laxatives and should be considered in patients whose motor control has deteriorated or where response to levodopa is variable.
6.7.1.2 Dysphagia
Dysphagia is not uncommon. Rarely recurrent aspiration pneumonia can complicate late stages of the disease. Patients benefit from access to a speech and language therapist to teach strategies to improve swallowing. Dysphagia is not typically levodopa-responsive and can deteriorate after deep brain stimulation. A dry oropharyngeal mucous membrane due to anticholinergic agents is one readily treatable cause of swallowing impairment.
6.7.1.3 Sialorrhea
This is a manifestation of reduced swallow frequency in IPD as opposed to excessive saliva production. Anticholinergics are effective, but most available agents are tertiary amines that enter the CNS and can impair memory or cause hallucinations in older patients. Quaternary amines do not penetrate the CNS and are preferable. Sublingual 1 % atropine can be used with some success. Injections of botulinum toxin into the salivary glands can be attempted. Pharyngeal weakness due to local toxin diffusion is a potential complication but is rarely encountered with dry mouth being a more common side-effect.
6.7.1.4 Orthostatic Hypotension
Orthostatic hypotension (OH) can cause significant morbidity and contributes to the risk of falling. Conservative measures such as increased fluid in-take, additional dietary salt, avoidance of hot baths and large meals and the use of compression stockings can help. More resistant symptoms can respond to the sympathomimetic midodrine, starting with 5 mg and titrating up to three doses of 10 mg a day if necessary. Fludrocortisone can be used, typically starting at 0.1 mg/day but supine hypertension can result from increased salt and mineralcorticoid ingestion. Elevation of the top of the bed to 30° at night may help by reducing renal mineralcorticoid production. OH can be aggravated by dopaminergic therapy (dopamine agonists in particular), dehydration and constipation.
6.7.1.5 Urinary Symptoms
Detrusor hyperreflexia predominates in IPD causing frequency, urge, nocturia and sometimes incontinence. In older male patients the picture may be mixed with prostatism and anticholinergics are ideally prescribed after bladder ultrasound to determine post void residual volume, avoiding exacerbation of pre-existing outflow obstruction. Equally important is the propensity of these agents to cause cognitive impairment in older patients with IPD, in particular the tertiary amines that cross the blood–brain barrier.
Trospium chloride is a quaternary amine that may have a better side effect profile although there is little trial data available addressing this issue. Reduction in late night fluid intake can help nocturia. In patients treated for OH nocturia can occur as a result of nocturnal pressure natriuresis secondary to supine hypertension.
6.7.1.6 Sexual Dysfunction
Sexual dysfunction is more commonly encountered in IPD than in the general population. Men with erectile dysfunction can be treated with agents such as sildenafil, however this can exacerbate OH. Female patients may report reduced libido and conversely hypersexuality can occur with dopamine agonist treatment and is particularly troublesome if associated with an impulse control disorder (discussed later).
6.7.1.7 Pain
Pain is not uncommon and can vary from uncomfortable paraesthesias to nonciceptive or neuropathic sounding pain. Patients can initially present with pain in a joint on the symptomatic side, typically a shoulder, probably due to hypokinesis and immobility. Adequate treatment and physiotherapy can improve this considerably. Some patients complain of pain down one side of their body or in an apparently radicular distribution, both of which will respond to levodopa suggesting a central dopamine deficit as the underlying cause. True radiculopathies from nerve root compression can also worsen in the ‘off’ state. Restless legs syndrome can be seen in association with IPD and can give rise to an aching discomfort in the legs at night that can improve with a low dose of a dopamine agonist taken at night.
6.7.1.8 Abnormal Sweating
The pathophysiology of abnormal sweating in IPD is unclear but Lewy body pathology involving the hypothalamus may be contributory. Sympathetic cholinergic fibers are the final common pathway that mediate the sweating response although dopamine would appear to play a role, as excessive sweating of the head and upper body can occur as an ‘off’ phenomenon, often in bed at night. Sweating can also occur in the context of dyskinesias, but is usually less prominent than the paroxysmal attacks of drenching sweats reported in ‘off’ periods. Other causes of excessive nocturnal sweating should be considered including thyrotoxicosis and latent tuberculosis infection.
6.7.2 Sleep Disturbance and Daytime Somnolence
Sleep disruption is common and multifactorial in IPD. Patients experience difficulty initiating sleep, fragmented sleep, REM sleep behavior disorder (RBD) and inversion of the sleep-wake cycle. RBD can predate the clinical onset of IPD sometimes by up to 10 years. Sleep disruption can exacerbate the excessive daytime somnolence that is both associated with the disease itself and dopaminergics.
Sleep disruption in IPD probably relates to degeneration of brainstem nuclei that regulate the balance between sleeping and waking states. The pedunculopontine and subcoeruleal nuclei are thought to play a role in maintaining the normal muscle atonia of REM that is lost in RBD. Involvement of nondopaminergic nuclei important in maintaining arousal including the raphe nuclei (serotonin), locus coeruleus (noradrenaline), the tuberomamillary nucleus (histamine) may account for daytime somnolence. The burden on bed-partners can be significant. Factors contributing to sleep disruption and therapies are given in (Table 6.5).
Table 6.5
Causes and treatment of sleep disturbance in Parkinson’s disease
Bradykinesia and rigidity | Can make it difficult to turn in bed to find a comfortable position. Contribute to difficulty initiating sleep or returning to sleep after an arousal. Some patients overcome this by using satin sheets and nightclothes to facilitate movement |
Restless legs syndrome (RLS) | Will respond to dopamine agonists and levodopa preparations given late at night. Treatment can be complicated by augmentation whereby symptoms become longer lasting, more severe and more extensive. It is important to ensure dyskinesias are not the cause of disturbed sleep as increased dopaminergic treatment will exacerbate this. Opioids, such as propoxyphene, can often suppress RLS and not cause augmentation |
Periodic limb movements in sleep | Periodic episodes of rhythmic extension of the hallux with dorsiflexion of the ankle, sometimes extending proximally to involve knee and hip flexors. Commonly associated with RLS and can also respond to dopaminergic drugs. Opioids can also be of benefit in resistant cases. Propoxyphene 65 mg late in the day before the onset of symptoms is usually effective. Start with a half-tablet, and titrate up to two tablets if necessary |
Nocturia | Common in this age group. Anti-cholinergics can help but may exacerbate vivid dreams or hallucinations. Sometimes responds to dopaminergic treatment. Rule out co-existing pathology with referral to urology for assessment where appropriate |
Vivid dreams | Are usually not disruptive to sleep but can be upsetting. Can resolve with a reduction in dopaminergic or anticholinergic drugs taken at night. Can be exacerbated by amphetamine metabolites of selegiline which should be taken early in the day. Low dose quetiapine, starting at 12.5–25 mg at night, can help if required |
Nocturnal hallucinations | Are associated with cognitive impairment in IPD and along with vivid dreams can respond to a low dose of quetiapine that can also improve insomnia due to its soporific effects. Donepezil 5–10 mg nocte can also be helpful |
REM sleep behavior disorder (RBD) | Semi-purposeful movements in sleep, typically as if kicking or fighting off an attacker. Occurs as a consequence of losing normal physiological paralysis during REM sleep. RBD is typically reported by bed-partners who should be questioned. A small dose of clonazepam, 0.25–1 mg at night, can be very effective. Melatonin, 3–12 mg at night, is an alternative when clonazepam exacerbates daytime somnolence and is generally well tolerated
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





