1
CHAPTER
![]()
Pathophysiology
Cheolsu Shin
The past decade has seen an explosion of the discovery of various genetic causes of many diseases, including epilepsy. With the rapid technological development in genomic sciences, many different approaches to finding genetic defects and variations underlying many disease states and epileptic conditions have been made available. The National Institutes of Health (NIH) supported a multicenter project to gather genomic and phenomic data on epileptic siblings or families that is expected to provide further rich soil for important discoveries. Furthermore, it seems that the time when individuals can have their entire genome sequenced for individualized management is approaching.
In this setting, the International League Against Epilepsy (ILAE) also proposed revised classification of seizures and epilepsies to incorporate the improved understanding of the pathophysiology of many seizures and epileptic conditions into the classification schemes (1). There have been many genetic defects and alterations identified that can lead to abnormal neuronal excitability. In some cases, it is easy to deduce the pathophysiology if the defect involves the molecular cascade underlying neuronal excitability. Ion channel or neurotransmitter receptor system abnormalities can easily be translated into abnormal physiology leading to an epileptic process. Other may not be that obvious, with seizure being the indirect result of the genetic abnormality. The bulk of the epileptic conditions still await further elucidation of their underlying pathophysiology.
There are two potentially separate processes that are involved in the epileptic brain. When a previously normal brain acquires the tendency for the occurrence of epileptic seizures, this is referred to as the epileptogenic process or epileptogenesis (2). When a seizure occurs in a brain that already possesses this tendency, this is referred to as an ictogenic process or ictogenesis (3). This may sound like a semantic argument, but there is ample experimental evidence that these two processes are different, although they may share some pathways and may also interact with and promote one another. Understanding the epileptogenic process may allow for early intervention in susceptible populations to prevent the eventual development of epilepsy. Better understanding of the ictogenic process may lead to better pharmacological or physiological management of the existing epileptic brain.
Since neuronal excitability is based upon the actions of various receptors and ion channels that occur at the level of the neuronal membranes and synapses, it is easy to explain some of the epileptic conditions given known molecular genetic alterations (4). If there are changes in the ion channels (sodium, potassium, calcium, chloride, etc.), then the resulting hyperexcitability may be obvious. If the excitatory neurotransmitter receptor system is hyperactive, or if the inhibitory neurotransmitter system is hypoactive, then hyperexcitability would be the likely consequence. However, the neuronal network is not that simple, as there are complex connections, feedback loops, presynaptic versus postsynaptic actions, etc., so that hypoactivity of the excitatory system controlling the inhibitory circuit may result in hyperexcitability.
There are also obvious anatomic abnormalities that cause epilepsy, such as brain tumors, tuberous sclerosis complex (TSC), cavernous hemangioma, or cortical dysplasia. Removal of the anatomic lesion leads to the abolition of the seizures. How these anatomic abnormalities cause the epileptic condition has not been fully elucidated.
In addition, neurons do not exist in a vacuum. They are supported by glial cells and are surrounded by the systemic circulation, including the blood–brain barrier. The glia do not just passively provide structural support for the neuronal network. They actively participate in regulating neuronal excitability and the understanding of their role in epilepsy is improving (5).
Many epileptic conditions may also entail involvement of inflammatory mechanisms. Some of these conditions are obvious, as in acute cases of meningitis or encephalitis that cause symptomatic seizures. Others may be more chronic, as in Rasmussen’s encephalitis. It is also likely that these inflammatory mechanisms are involved in seizure-induced brain injury and epileptogenesis. Recently, more awareness of the immune mechanism underlying some of the epileptic processes was made when autoimmune antibodies were identified that are directed against neuronal tissues causing inflammatory reactions and epilepsy (6).
CELLULAR PHYSIOLOGY
Epilepsy ultimately involves a neuronal population and as such is mechanistically based upon excitable membrane physiology. Neuronal excitability is based upon the regulation of ion channels that are selectively permeable to specific ions that make up the intracellular and extracellular milieu. Membrane voltage is determined by the Nernst equation that incorporates the conductances of the select ion channels.
![]()
Because of the differential ionic concentrations between the intra- and extracellular spaces, each ion-selective channel will have an equilibrium potential (reversal potential) that balances the chemical gradient and the electrical gradient. Whichever ion channel is open will contribute to or dominate the membrane potential. When the potassium channel is open, the membrane will hyperpolarize. Sodium channel opening will lead to depolarization. Chloride channel opening will not change the resting membrane potential much, as its equilibrium potential is near the resting potential, but its opening will shunt the currents to reduce the depolarizing or hyperpolarizing force from other channel openings. Calcium channel opening provides a much stronger and longer depolarization force, by virtue of being a divalent cation with double the current of monovalent sodium ions and by having a much steeper concentration gradient than sodium. In addition, calcium ion can trigger many cascades of intracellular signaling that can lead to neuronal plasticity. These neuronal membrane ionic conductances would work only if the ionic milieu is maintained. Ionic pumps utilizing adenosine triphosphate (ATP) energy source (eg, Na/K ATPase) will restore the ionic concentration gradients, as the neuronal activity continually causes ion transfers across the membrane through ion channels.
Many of the neurotransmitter receptors may function as ion channels. The gamma-aminobutyric acid (GABA)-A receptor is a chloride ion channel with allosteric modulation by benzodiazepines and barbiturates. Therefore, GABA-A activation would be inhibitory by keeping the membrane potential near the resting membrane potential. However, in the special situation of the developing fetal brain, the ionic gradient is maintained in such a way that GABA-A activation may actually depolarize the membrane. The GABA-B receptor is a metabotropic receptor that is indirectly linked via a G-protein to the potassium channel, which hyperpolarizes the membrane. Excitatory amino acid receptors of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or kainate act as gates of sodium ions and therefore depolarize the postsynaptic membrane. The N-methyl-d-aspartate (NMDA) subtype of glutamate receptors are channels that also allow calcium entry. They require not only the binding of glutamate, but also depolarization, usually provided by the activity of AMPA glutamate receptors. This provides for an intense synaptic activity, which in turn allows calcium entry that can then trigger steps leading to neuronal plasticity. When the activity is too intense, the system may break down and neuronal injury and subsequent epileptogenesis may ensue.
GENETIC EPILEPSY
There are many epilepsy syndromes that have identified genetic defects. There are several varieties of syndromes now described, the review of which is beyond the scope of this chapter, but there are many reviews that describe the molecular details (7,8). Many of these defects have obvious relevance to the epileptic condition with molecular abnormalities in the system directly underlying neuronal excitability. Some of these mutations have been validated in experimental systems to be the likely direct cause for the epileptic condition. Other mutations are not always obviously related or validated. In addition, the genetic abnormalities do not always translate into a uniform phenomenon throughout the brain. In the past, it may have been a safe assumption that if the epileptic condition is genetically caused, it must be a generalized epileptic condition. It is now known that many genetically identified epilepsies are focal, as in autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE; nicotinic cholinergic receptor defect) and autosomal dominant partial epilepsy with auditory features (ADPEAF; leucine-rich glioma inactivated 1 (LGI1) mutation). In severe myoclonic epilepsy of infancy (SMEI; Dravet’s syndrome; SCN1A sodium channel mutation), there are cell-specific subunit expressions to GABAergic inhibitory neurons in the cortical and hippocampal regions, whereby loss of function in the sodium channel would lead to loss of inhibition and therefore hyperexcitability (9). Therefore, abnormal gene expression may be directed to an area of the brain and to a subpopulation of neurons, be it excitatory or inhibitory, and may underlie focal hyperexcitability, leading to focal epilepsy syndromes.
There are also many genetic conditions where epilepsy is a prominent feature of the disorder, such as TSC. Some genetic conditions cause neuronal migrational abnormalities that are then associated with epileptic processes (eg, bilateral periventricular nodular heterotopia syndrome with X-linked dominant mutation of filamin-1 gene). Understanding how epilepsy develops in these genetic conditions may also shed light on other epileptic processes.
In TSC, epilepsy is a prominent feature (10). Infantile spasms occur frequently in this condition and focal seizures may originate from the cortical tubers. In many cases, the seizures are refractory to medications. Resection of the cortical tubers identified as the focus of seizures frequently leads to many seizure-free years, thus implicating tuber formation in the pathogenesis of epilepsy (11). The TSC protein complex is a negative regulator of the mTOR signaling pathway, and the TSC protein complex mutation leads to abnormal activation of this pathway. This leads to the formation of various tumors, including the subependymal giant cell astrocytoma (SEGA). Rapamycin inhibits the mTOR pathway and it has been shown to be effective in the treatment of SEGA. However, in the already established epileptic condition of TSC, rapamycin is not proven to be effective in controlling seizures. Since there is an overabundance of glia in the tuber along with giant cells, it is likely that the epileptogenesis occurs through mechanisms related to glial dysfunction. As discussed in the following sections, glial proliferation and abnormal glial morphology can lead to shrinkage of the extracellular space and reduced buffering capacity for potassium and glutamate, all promoting abnormal neuronal excitability in and around the cortical tubers.
AUTOIMMUNE EPILEPSY
Recent report suggests that there may be another mechanism underlying epilepsy, especially drug-resistant epilepsy. Many epilepsy centers evaluate patients with pharmaco-resistant epilepsy and evaluate them for a resective surgical option. In cases of normal high-resolution anatomic imaging, there may be a microscopic focus of abnormal circuitry. In these cases, functional imaging and invasive electrophysiology are used to locate the source of the epileptic process. However, there are many situations where there does not seem to be a good surgical solution.
At least in some of these cases, there may be additional pathophysiologic mechanisms to be considered. Seizures may occur in obvious cases of inflammatory processes, such as viral encephalitis, including herpes simplex encephalitis that preferentially affects the temporal lobes. Cerebritis related to systemic immune dysfunction, such as lupus, can be associated with seizures. Rasmussen’s encephalitis that may involve a hemisphere, mostly in young people, is a well-known entity with likely participation of cell-mediated immune mechanisms. There are well-recognizable situations of paraneoplastic limbic encephalitis.
Nevertheless, there are patients who do not have any significant encephalopathy but have refractory seizures. In one study of these cases, neuronal autoantibodies were found mostly directed to voltage-gated potassium channel complex, glutamic acid decarboxylase (GAD) 65, collapsing response mediator protein (CRMP) 5, Ma, NMDA-receptor, and neuronal nicotinic ganglionic acetylcholine receptor (6). There were imaging abnormalities, some of which were subtle and initially not noticed. A few patients had a neoplasm. Immunotherapy consisting of high-dose intravenous methylprednisolone, intravenous immunoglobulins, or combinations including plasma exchange or cyclophosphamide resulted in seizure freedom in two-thirds of the patients. Eight of 18 responders (44%) became seizure-free within 12 weeks of initiation of immunotherapy. This study raises many questions regarding a major subset of patients with pharmacoresistant epilepsy. In the appropriate clinical setting, it is important to consider immunologic mechanisms as a cause of medical intractability. Eventually, controlled clinical trials and studies of basic immune mechanisms should lead to a more rational basis for alternative therapy options.
ROLE OF ASTROGLIA IN EPILEPTOGENESIS
Although the final manifestation of epileptic seizures comes through as synchronized abnormal neuronal activity, the role of astroglia cannot be ignored. The glia is not just the supporting structural framework for the neuronal network, but it is an integral part of the homeostatic milieu that is necessary for neuronal integrity and function. Furthermore, there is active interaction between neurons and glia during normal neurotransmission. Any perturbation of these neuron–glial interactions leads to pathology, including hyperexcitability that may lead to epileptogenesis. In addition, the glia, along with the vasculature, forms the blood–brain barrier that insulates the brain from many systemic influences and insults. When the blood–brain barrier is disrupted, inflammation may ensue, and with all of its injurious components, the process of epileptogenesis may begin (12). Autoimmune epilepsy discussed earlier may well be an example of such a process.
Even focal epileptic conditions such as mesial temporal lobe epilepsy have been shown to have severe gliosis in the hippocampus, leading to terminology such as Ammon’s horn sclerosis or mesial temporal sclerosis (MTS). Obviously, where there is excessive gliosis and absence of neuronal elements, seizures do not originate from that region. However, seizures may be triggered from the adjacent areas where the gliosis may be impacting neuronal function.
Since glia cells regulate the ionic milieu of neurons, they can alter neuronal excitability by affecting water flow and ionic buffering, including potassium. This can change the extracellular space and the ionic concentrations around the neurons. Shrinking the extracellular space will increase neuronal excitability due to higher extracellular potassium concentration and possible ephaptic neuronal synchronization. Hypo-osmolar situations (water intoxication or severe hyponatremia) swell the cells and thereby shrink the extracellular space, and these situations may cause seizures. Aquaporin-4, the glial water channel and the inward rectifying potassium channel (Kir) may be part of the glial control mechanism in this respect, and their dysfunction may lead to hyperexcitability (13).
Glia also take up glutamate released from the glutamatergic excitatory terminals to limit the spread of the neurotransmitter to extrasynaptic sites or adjacent synapses. In addition, glutamate released by the synapse can activate the glial metabotrophic glutamate receptors that increase the glial intracellular calcium. This elevation of intracellular calcium then releases glutamate to the extracellular space. This constitutes bidirectional neuro-glial communication. Similar bidirectional communication may occur with other neurotransmitters. Whether this bidirectional communication is physiologically important is unknown. However, in pathologic situations where glial proliferation takes place, the extracellular space may already be reduced, and glial release of glutamate may promote synchronized epileptic activity in the surrounding neuronal network, as evidenced by in vitro experiments (14).
The blood–brain barrier, along with the microvasculature of the brain, is formed by glia. Glia maintain the tight junctions between the endothelial cells and facilitate or filter the transfer of a variety of molecules from the systemic circulation to the brain parenchyma and vice versa. As such, glia would be part of any process that would allow immune system access to the brain parenchyma. In cases of autoimmune epilepsy, neuronal autoantibodies may be accessing the brain parenchyma, somehow via participation of the glia. In cases of injury to the brain, be it trauma, infection, or ischemic insult, there is reactive inflammation that seeks to repair the damage. Since so many complex cascades of microglial and other reactive elements participate in these repair processes, it may be very difficult to tease out which parts would end up promoting epileptogenesis. This endeavor at the basic science level may be critical in elucidating the mechanisms of epileptogenesis in acquired epileptic conditions, so that one can devise ways to intervene to prevent the development of epilepsy. There is accumulating evidence for the pathogenic role of glia underlying hyperexcitability in epilepsy through synchronizing glial neurotransmission, by controlling the extracellular space volume and ionic milieu, and participating in inflammatory processes (5).
MESIAL TEMPORAL SCLEROSIS
The temporal lobe appears to be the most frequent anatomic site for epileptogenesis. In particular, the hippocampal formation and its surrounding structures of subiculum, entorhinal cortex, and parahippocampal gyrus appear to be particularly involved in the epileptic process in humans. Anatomic connections of the hippocampal circuitry are well known (Figure 1.1). The entorhinal cortex provides input to the dentate granule cells via the perforant path; dentate granule cells then innervate the CA3 pyramidal neurons via the mossy fibers. The CA3 pyramidal neurons innervate the CA1 pyramidal neurons via the Schaffer collaterals, and subsequently, CA1 pyramidal neurons provide output to the subiculum and through the fornix to the extrahippocampal areas. These synapses are excitatory and therefore may serve as an amplifier and, along with inhibitory interneurons, as a fine-tuning circuit for new memory formation. Because of the compact layers of neurons and ample excitatory synapses in these pathways, the hippocampus is one of the most susceptible structures in the brain for neuronal damage from a variety of insults (ischemic, traumatic, inflammatory) and from excessive seizure activity. As a result, the pathological entity of MTS has been well recognized and it is now more easily detected in vivo using high-resolution magnetic resonance imaging (MRI) scans. Many experimental models have been used to study the process of epileptogenesis in the hippocampus in vivo and in vitro, with much insight gained into the pathogenesis of epilepsy from this region of the brain (15).
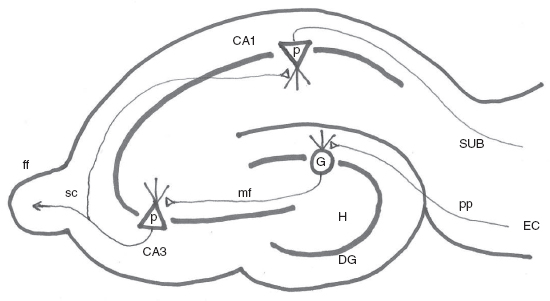
FIGURE 1.1 Schematics of Hippocampal Circuitry. Entorhinal cortex (EC) innervates the dentate granule cells (G) via the perforant path (pp). Granule cells send mossy fiber (mf) that innervates the CA3 pyramidal neurons (p). CA3 pyramidal neurons then send Schaffer collateral fibers (SC) that innervates CA1 pyramidal neurons, which in turn send fibers to subiculum (SUB) and thus completing the trisynaptic excitatory connections.
Abbreviations: Fimbria-fornix (ff); dentate gyrus (DG); dentate hilus (H)
![]()
These epileptogenic processes appear to involve new synapse formation as part of plasticity of the brain, albeit maladaptive. Protein synthesis inhibition can curb mossy fiber sprouting and block epileptogenesis. Which molecular pathway is critical in mediating this maladaptive plasticity is obviously an area of active research in many laboratories. Of many potential molecules, brain-derived neurotrophic factor (BDNF) pathway may be a very promising candidate (17).
BDNF is one of many signaling molecules in the brain that triggers a cascade of intracellular events that underlie neuronal plasticity. It belongs to the nerve growth factor (NGF) family that promotes the survival of neurons and the growth of their neurites and processes. It binds to a receptor, TrkB, that phosphorylates tyrosine moieties in the protein, leading to activation of intracellular signaling cascades. In many animal models, seizures induce increased BDNF expression that promotes activation of its TrkB receptor in the mossy fiber pathway. Experimental manipulation of BDNF or TrkB expression and activation appear to have a profound effect on the development of the epileptic condition. If the BDNF/TrkB activity is increased, then the epileptogenic process is enhanced. If, on the other hand, the BDNF/TrkB expression is blocked, then the epileptogenic process is attenuated. This BDNF/TrkB pathway may provide an important avenue of clinical application aimed at blocking epileptogenesis in situations of brain injury that are highly likely to lead to epilepsy.
PATHOPHYSIOLOGY OF PRIMARY GENERALIZED EPILEPSIES
Classic childhood absence seizures with 3 Hz spike and wave discharges have been better characterized in terms of pharmacology and pathophysiology than most other forms of epilepsy. Ethosuximide has been used as a very specific medication for this form of epilepsy with its T-calcium channel blocking activity identified as a mechanism of its antiepileptic property.
In terms of the network concept of epilepsy, the centrencephalic theory had been initially proposed with the reticular thalamus as the pacesetter. Subsequently, the corticoreticular theory postulated the involvement of the cortex in thalamocortical excitability. Some animal model studies suggest a cortical focus that entrains the cortex as well as the thalamus to generate self-sustaining spike wave discharges (18). Whether the animal model accurately predicts human absence epilepsy is unclear. Nevertheless, these studies suggest a basis for the absence epilepsy involving the cortex as well as the thalamus in a circuit that includes ion channels and neurotransmitter receptor systems that support the reverberating excitatory discharges (Figure 1.2).
The reverberating corticothalamic connections are modulated by the GABAergic innervation. GABA-B receptor activation on the thalamic relay neurons (T) provides the hyperpolarization needed for the de-inactivation of the T-calcium channel so that abnormal 3 Hz spike and wave discharges can be sustained. GABAergic interneurons (G2) may inhibit the GABA-B projection (G1) by GABA-A inhibition or may directly inhibit the relay neuron (T). Drugs effective in absence epilepsy, ethosuximide (ETX) and valproate (VPA), inhibit T-calcium channels. Benzodiazepines (BDZs) through the GABA-A mechanism.
As mentioned, the low threshold T-calcium channel is an integral part of the thalamocortical network communication (19). Its electrophysiological properties may explain some of the pharmacological effects of the GABA receptor system on absence seizures. T-calcium channels have a similar property to the voltage-gated sodium channels, in that once it is opened with depolarization, it becomes inactive and closed until it is de-inactivated. That de-inactivation requires a significant degree of hyperpolarization beyond the resting membrane potential. Many channels contribute to the return from depolarization, including the GABA-A receptor-coupled chloride channel. More recently, hyperpolarization-activated cyclic nucleotide-gated (HCN) channels have been implicated in the oscillation of thalamocortical circuitry (20). Like the T-calcium channel, the HCN channel is inactivated by depolarization and requires hyperpolarization for de-inactivation. Its reversal potential is –25 to –40 mV so that it may not provide the depolarization offered by the T-calcium channel, but may underlie the physiological as well as pathological oscillatory network behavior of the thalamocortical circuitry.
Thalamic relay neurons receive excitatory inputs from the cortex and may transmit a similar output back to the cortex, activating the T-calcium channel once. When the return volley of corticothalamic projections depolarizes the thalamic neuron, the T-calcium channels (and the HCN channels) are still in the inactive state, so that the oscillation is dampened and restrained in the physiological state. However, if there are hyperpolarizing inputs, that could change the delicate physiological balance. GABA neurons in the reticular thalamus also receive the cortical projections and in turn project to the relay neurons to modulate the activity. Although the GABA-A receptor function stabilizes the relay neurons, GABA-B receptors provide the hyperpolarizing force by activating the potassium channel that is linked to G-protein. In contrast to the GABA-A receptor-chloride channel with its reversal potential of –60 mV, which is near the resting membrane potential, the potassium channel reversal potential is usually –80 to –100 mV, which allows de-inactivation of T-calcium channels, resetting them so that they are ready to open. Thus, the next corticothalamic volley will result in a powerful depolarizing force, leading to the reverberation of 3-Hz spike and wave discharges. Therefore, the pharmacological correlates of this circuitry would predict that attenuation of T-calcium channel activation would block the hyperexcitable reverberation, and indeed, ethosuximide and valproate, both T-calcium channel blockers, are effective against absence seizures.
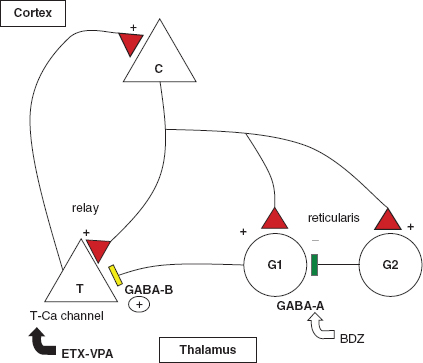
FIGURE 1.2 Functional anatomy of absence seizure. The reverberating corticothalamic connections are modulated by the GABAergic innervation. GABA-B receptor activation on the thalamic relay neurons (T) provides the hyperpolarization needed for the de-inactivation of the T-calcium channel so that abnormal 3 Hz spike and wave discharges would be sustained. GABAergic interneurons (G2) may inhibit the GABA-B projection (G1) by GABA-A inhibition or may directly inhibit the relay neuron (T). Drugs effective in absence epilepsy, ethosuximide (ETX) and valproate (VPA) inhibit T-calcium channels. Benzodiazepines (BDZ) would work through the GABA-A mechanism.
![]()
It is also known that benzodiazepines that facilitate GABA-A receptor activation also block absence seizures. This action may be due to direct inhibition of both cortical and thalamic neurons, but it is also possible that GABAergic neurons in the reticular thalamus may also inhibit the GABAergic neurons that provide GABA-B projection to the thalamic relay neurons. As such, GABA-B antagonist in this scheme will be an effective antiabsence drug, whereas GABA-B agonist would more likely promote absence epilepsy.
MECHANISM OF ICTOGENESIS
The discussion so far has mostly addressed how epilepsy develops in a previously normal brain, ie, epileptogenesis. This is a critically important issue to understand in order to prevent the occurrence of epilepsy. However, practically, it is important to manage the patients who already have gone through the process of epileptogenesis and are now having recurrent spontaneous unprovoked seizures. Of course, pharmacotherapeutics have come a long way since the past century with many new antiepileptic agents available and many new ones being investigated. These pharmacotherapeutics have been geared toward molecular targets of ion channels and neurotransmitter receptors, with agents that target sodium channels, potassium channels, GABA-A receptors, GABA transaminase inhibitors, and AMPA receptors.
More recent electrophysiological studies have shed light on a very interesting concept of how seizures start in the already epileptic brain (21). Newer understanding of this perspective may lead to novel therapeutic approaches, both pharmacologically and electrophysiologically. Ever since the EEG was discovered, the frequency range of brain electrical activity recorded has ranged between 1 Hz and 70 Hz, initially limited by the electronics of the amplifiers and the output of the tracings onto paper using an array of ink-pen printing mechanism. Now, in the presence of all digital EEG interface, there is no reason to be limited in frequency range in analyzing brain electrical activity. Experimental models of epilepsy have already studied some of these high-frequency activities, and recent advances in digital electronics and computing power have enabled similar studies in humans who are undergoing epilepsy surgery in cases of medically refractory epilepsy.
There are many situations where the resection of a lesion, be it a tumor, cavernous hemangioma, or gliotic scar tissue, leads to seizure freedom in a previously refractory epileptic patient. In many cases, however, there has been no anatomic abnormality on neuroimaging studies with the need to resort to more invasive intracranial EEG monitoring in an effort to identify an epileptic focus for resection. Even then, it may not be possible to identify a focus, or resection of the putative focus may not achieve seizure freedom, leading to the conclusion that the epileptic focus was not fully resected.
In this setting, there is some emerging evidence that the identification of ultra-fast bursting activity (fast ripples) may represent the electrophysiologic biomarker for the epileptic focus. Resection of the brain regions with these discharges may lead to improved surgical outcome (22). These fast ripples, 250 Hz to 500 Hz bursting activity, have been recorded interictally during intraoperative electrocorticography and they seem to be present in the epileptogenic tissue. If the cortex where fast ripples are recorded is resected, seizure freedom is more likely. This finding was seen in a retrospective study with the need for further prospective studies to verify this hypothesis (22). This study has some weaknesses, but still may point toward an important direction for the future investigations (23).
The fast ripple is a pathological high-frequency oscillation (pHFO) that may indeed be a promising biomarker for localizing epileptogenic focus. As presented in reports from the conference on high-frequency oscillations (HFOs) in cognition and epilepsy (24), there is now much work ongoing in this field regarding normal and pathological significance of these high-frequency activities recorded from the brain, cortex, hippocampus, and subcortical structures. It appears that there are normal HFOs that occur in various brain areas, sometimes associated with different activities or states and at other times seemingly randomly. Underlying molecular and network mechanisms are beginning to be dissected. Whether the pHFOs contribute to the pathogenesis or are simply biomarkers of the epileptogenic zone or both is unclear. Regardless, this area of investigation holds great promise in furthering the understanding of molecular and circuit network behavior underlying the epileptic process.
There are neurons that physiologically fire repetitively in oscillation. It is hard to imagine, however, that a single neuron can be the starting focus of an epileptic process involving a large brain area with innumerable neurons. A network of pathologically active circuitry may be more likely the underlying process. In the thalamocortical reverberating circuitry, hyperactive low threshold calcium channel (T-calcium channel) could entrain the network into the epileptic population behavior of 3 Hz spike and wave discharges recorded using scalp EEG and resulting in the absence seizure. For focal seizures, there is no intrinsic reverberating circuitry that can be entrained. However, in the injured area of the brain, or malformed or disorganized neuronal population, one may envision molecular and local alterations that may form the basis of some reverberation. Excitatory synapses that are hyperactive due to maladaptive plasticity may contribute. Also, ephaptic mechanisms may contribute in compact layers of neurons oriented in a single direction as in the hippocampus. Gap junctions may synchronize inhibitory neurons to hypersynchronize the network of bursting neurons. Gliosis and abnormal glial environment also contribute by shrinking the extracellular volume and reducing the buffering capacity for any spilled excitatory glutamate neurotransmitter and increased potassium ions. These all combine to form islets of small groups of neuronal circuits that may cause HFOs that are pathologic. These groups of small populations of neurons may not be able to launch an epileptic seizure on their own. But, given additional provocative factors, they could be recruited to synchronize in larger groups that may eventually be entrained to generate a clinical seizure phenomenon. Once a seizure is generated through the network, it may leave behind an engram that subsequent seizure events may follow to result in a stereotypic electrographic and behavioral epileptic pattern.




