CHAPTER 37 Pathophysiology of Subdural Hematomas
Chronic subdural hematoma (CSDH) is frequently encountered in neurosurgical practice and occurs at a rate of 1 to 2 per 100,000 per year. Nonetheless, there has been ongoing debate over the fundamental pathophysiologic mechanisms of the development, evolution, and recurrence of CSDH. Virchow1 in 1857 first described pachymeningitis haemorrhagica and ascribed the condition to dural inflammation; however, by the early 20th century, the traumatic nature of CSDH was established and widely accepted. During the subsequent century, through ultrastructural analysis of subdural membranes and various components of CSDH fluid, a complex pathophysiology has evolved.
Pathophysiology of the Development of Chronic Subdural Hematomas
Grossly, CSDHs may vary in color from clear yellow to dark purple and in consistency from thin liquid to semisolid. A thin, often translucent inner membrane and a thicker outer membrane often encapsulate the hematoma.2,3 The contents of the hematoma and the histology of the outer membrane have been the focus of investigation on the mechanisms by which CSDHs develop and expand.2–6
In 1932, Gardner first proposed the osmotic gradient theory as the predominant pathophysiology of CSDH. He postulated that the increased protein content in CSDH fluid causes ingress of fluid as a result of increased oncotic pressure.7 Although CSDH fluid contains high levels of protein and lipid, Weir showed CSDH fluid to be isosmotic to both blood and cerebrospinal fluid.8 Instead of an osmotic force pulling plain water into the CSDH, microscopic examination of fluid from CSDHs of any age reveals fresh erythrocytes, thus indicating that clinically silent rehemorrhage or progressive leakage of fresh blood into the CSDH is ongoing.9,10
The probable source of rebleeding is the CSDH membranes. These membranes consist of blood vessels, eosinophils, smooth muscle cells, fibroblasts, and myofibroblasts supported by a matrix of collagen and elastin.4,5,11 The membranes arise from cleavage of the dural border layer, which results in dural border cells on both sides of the hematoma cavity. Some elements of both the inner and outer membranes resemble this dural border layer; however, the presence of blood vessels and eosinophils and attenuation of the remaining cells set a CSDH membrane apart.4
Blood vessels are absent in the normal dura-arachnoid interface. Neovasculature is abundant, but just in the outer CSDH membrane. Abnormal dilated sinusoids measuring as large as 1000 µm, with an incomplete basement membrane and attenuated endothelial cells, share the outer membrane with rapidly growing microcapillaries. Both vessel types are composed of endothelial cells with irregular surface because of numerous pseudopod-like structures extending into the vascular lumen.6 Erythrocytes and platelets in various stages of degeneration are frequently found deposited in the perivascular space.4,6 These sinusoids contain gap junctions as large as 8 µm, sufficient to allow leakage of plasma and even red blood cells into the hematoma cavity.12
Inflammatory mediators present in CSDH fluid may potentiate chronic rebleeding of the fragile neovasculature. Kallikrein, bradykinin, and platelet-activating factor (PAF) have all been identified at significant levels in CSDH fluid. These inflammatory mediators stimulate vasodilation, increase vascular permeability, prolong the clotting time, and release tissue plasminogen activator (t-PA) from endothelial cells.13–15 Other work has focused on disturbances of the prostaglandin system (local and potentially systemic) as possible components in the pathophysiology.16
Eosinophil degranulation in the outer membrane may be the source of the fibrinolytic factors and inflammatory mediators causing local coagulopathy and cell destruction in the CSDH. An abundance of eosinophils in the outer membrane has been noted for decades.17,18
Evolution of Chronic Subdural Hematomas
As early as 1826, Bayle proposed that repeated bleeding episodes cause the ongoing expansion of CSDHs.1 Decades of clinical experience and modern technologies—including computed tomography, magnetic resonance imaging, advanced microscopy, molecular biology techniques, and other innovations—have further supported his theory. In an early computed tomographic study, Bergström and colleagues19 showed that acute subdural hematomas undergo a predictable loss of attenuation over time, but that some CSDHs return to high attenuation. They postulated that this change reflects an instance of sudden or chronic rebleeding as predicted by Bayle.19
As discussed previously, the presence of defective neovasculature certainly potentiates the process of rebleeding and absorption of plasma proteins into the subdural space, but this does not fully explain the growth of CSDHs. Normal hemostatic mechanisms should halt the process long before the CSDH reaches a clinically significant size.15,19,20 Instead, considerable evidence supports the presence of a localized coagulopathy within the CSDH.
Labadie and Glover21 analyzed fluid from two recurrent CSDHs. They found accelerated clot formation by the partial thromboplastin time and normal formation by the prothrombin time; however, the clots formed were structurally defective in comparison with controls. They postulated that the clots within CSDHs degrade rapidly, a supposition supported by high levels of fibrin degradation products (FDPs) and low mean plasminogen in their fluid samples.21 FDPs inhibit coagulation, platelet aggregation, and fibrin polymerization and promote the activity of t-PA.22
Subsequent studies have identified lower levels of all coagulation factors in CSDH fluid than in plasma. Factors II,V, VII, VIII, and X are disproportionately depleted.23 These findings reflect a phase of accelerated fibrinolytic activity after the rapid and defective clot formation. The end result is a milieu of anticoagulant proteins (chiefly FDPs) and depleted coagulation factors.2,21–24
The source of this accelerated fibrinolysis may be t-PA. t-PA transforms plasminogen into plasmin, which, in turn, degrades fibrin to fibrin split products (Fig. 37-1). Investigations by Ito and associates25 found a threefold increase in t-PA in the outer membranes of CSDHs as opposed to the dura. Weir and Gordon24 corroborated Ito and colleagues’ work by showing increased t-PA and decreased α2-antiplasmin (an inhibitor of plasminogen activation) in CSDH fluid.
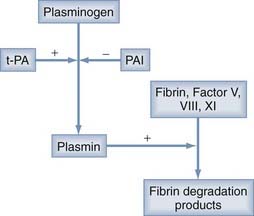
Other authors have suggested that the PAF derived from lysis of red blood cells may stimulate the synthesis and release of t-PA, as well as induce chemotaxis of inflammatory cells to the CSDH membranes. They found increased levels of PAF in CSDH fluid and elevated plasma levels of PAF in patients with CSDH versus healthy volunteers. The latter observation may suggest a systemic predilection to the development of CSDH.15
In measurements of CSDH fluid from 23 patients, Lim and coworkers22 found a correlation between the ratio of t-PA–to–plasminogen activator inhibitor (PAI) and hematoma size. These authors postulated that PAI may interrupt the hyperfibrinolysis to slow the growth of CSDHs or promote gradual spontaneous resorption.22
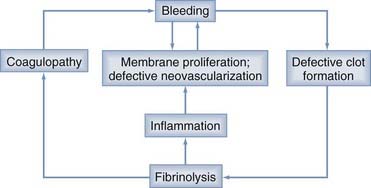
Localized coagulopathy and defective clot formation within the CSDH completes the cycle of bleeding, membrane formation, defective vascularization, fibrinolysis, coagulopathy, and rebleeding (Fig. 37-2).3,22






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


