Extracellular matrix destabilization occurs along with cellular dysfunction. The concentration of several matrix metalloproteinase (MMP) proteins are elevated post TBI in the brain tissue, CSF and blood samples of patients with poor neurological outcomes [36, 37]. As glial cells in the area of injury die, they form scars that may inhibit the survival of the remaining neurons and function of the local neural network.
The secondary injury also affects the cerebral vasculature and includes hypoperfusion, vasospasm and hypoxia. Injured brains are susceptible to hypoxic-ischemic states due to dysfunctional cerebral autoregulation and commonly encountered post traumatic vasospasm, both of which may contribute to further neurologic deterioration [38]. However, tissue perfusion and oxygenation does not always correlate with the severity of injury suggested by computed tomography or ICP measurements [39]. At the tissue level, angiogenesis occurs post injury, and is mediated by both the resident mature endothelial cells via capillary outgrowth but also by bone marrow or peripheral endothelial progenitor cells (EPC) arriving within 24 h in either a primary neovascular or supportive role [40].
Intracranial Dynamics
Brain edema, which classically peaks approximately 48–72 h after the initial trauma, is the primary target for neurointensive care. Edema results from both primary cellular death and tissue osmotic dysregulation. The severity of edema is currently monitored clinically by neurologic clinical exam, neuroimaging and intracranial pressure monitoring, the latter of which is used to guide therapy for adequate perfusion and oxygen delivery to the brain. Direct brain tissue oxygen tension monitoring and microdialysis may also be used along with intracranial pressure monitoring as multimodal therapy to optimize cerebral blood flow, oxygen and energy substrate delivery [41].
Three volumes comprise the brain and include the 1300 mL of brain parenchyma, 110 mL of blood and 65 mL of cerebral spinal fluid (CSF) [42]. The Monro-Kellie doctrine describes the intracranial dynamics following injury. Change in one of the three volumes must be compensated by the reduction in another component. Without volume changes, the ICP, which is normally at or below 10 mmHg, rises [43].
The brain parenchyma is mostly water, which is not compressible [44]. Intracranial blood is two-thirds venous and one-third arterial and is controlled via autoregulation and is influenced by arterial pressure, PO2 and PCO2. Cerebral perfusion pressure (CPP) defined as mean arterial pressure minus ICP is used as a surrogate for cerebral blood flow, and 50–70 mmHg is the clinical range recommended for optimal blood and oxygen delivery to the brain. CSF is produced at 10–20 cc/hr, and is taken up through arachnoid villi and plays an important role in the transportation of and clearance of metabolites.
Following traumatic brain injury, volume derangements occur to each of the three brain volumes. The brain parenchyma volume increases due to vasogenic and cellular edema [45]. In vitro studies suggest that mechanical stretch can activate cation channels in astrocytes that exacerbate cytotoxic edema [46]. Up to one-third of TBI patients have abnormal cerebral autoregulation. As the result of the loss of cerebral autoregulation, cerebral blood flow and capillary hydrostatic pressure increases, exacerbating edema and ICP. CSF circulation and clearance of metabolites is also impaired following TBI.
Clinical Neurointensive Care
The current management of the injured brain following TBI still remains largely supportive with much of the focus on optimizing cerebral perfusion by managing cerebral edema and intracranial pressures [5, 47]. Clinical care guidelines have been developed and are associated with improved outcomes when used by critical care teams [48–50]. However, these practices lack robust randomized control trials. Currently, the management strategy escalates in intensity in a tiered fashion with first tier treatments typically including sedation, establishing an ICP threshold, cerebral perfusion monitoring, neuromuscular blockade, CSF drainage and hyperosmolar therapy [51]. Second tier treatments include hyperventilation, barbiturates for pharmacological coma with electroencephalogram monitoring for burst suppression, hypothermia and surgical decompression.
First tiered therapy begins with sedation, which is a combination of anesthetics and analgesics. Propofol is a commonly used anesthetic used for quick clearance required for frequent neurological tests but can cause myocardial depression as part of an infusion syndrome and may not reduce the cerebral ischemic burden [52]. Norepinephrine and phenylephrine are commonly used pressors to maintain adequate cerebral perfusion pressure because they have the least effect on cerebral vasomotor tone, but overaggressive hypertension may increase the risk of acute respiratory distress syndrome [5]. Fever increases the cerebral metabolic burden and increases the ICP and is treated aggressively with a low index of suspicion for infection and atelectasis. Hyperosmolar therapy includes Mannitol, which is administered at 0.5–1 g/kg and produces effect within 15–30 min. This can be administered every 6 h to a target serum osmolaritiy of 310–320 Osm/L. In addition to lowering the intracranial pressure, Mannitol also has been shown to improve cerebral blood flow (CBF) [53]. 23 % hypertonic saline can be used for hyperacute ICP elevations and for herniation syndromes and can reduce the ICP by up to 50 % within minutes and produce a durable response over hours. Sodium chloride and sodium acetate can be used as a mixture to minimize hyperchloremic metabolic acidosis.
Second tier options include barbiturates and surgical decompression. Barbiturate coma reduces the cerebral metabolic rate as well as the ICP but also has numerous systemic risks, including hypotension, hypocalcemia, hepatic renal dysfunction, sepsis, and ileus. Furthermore, the long term outcome for barbiturate coma is unknown [54, 55]. The use of mild induced hypothermia (body temperature between 32–35 °C) has produced mixed results with some studies suggesting no benefit while others suggesting modest benefit [56]. Hypothermia does exacerbate electrolyte disorders, arrhythmia and infections. The DECRA (Decompressive Craniectomy in patients with severe traumatic brain injury) trial examined outcomes for patients undergoing decompressive craniectomies for elevated intracranial pressures. Although decompression lowers intracranial pressure and decreases length of stay, the investigators cited worse long term outcomes. However, the trial did not include a significant population undergoing craniectomies, namely those with space occupying hematomas or those undergoing unilateral craniectomies [57]. The international multicenter RESCUEicp (Randomised Evaluation of Surgery with Craniectomy for Uncontrollable Elevation of Intra-Cranial Pressure) has been designed to compare surgical decompression versus medical management alone.
Experimental therapies aim to improve the monitoring and therapeutic response to the post injured brain. A controversial issue in neurocritical care is in regard to the practice of directed therapy to maintain ICP levels below 20 mmHg. The multicentered randomized Benchmark Evidence from South American Trials: Treatment of Intracranial Pressures (BEST TRIP) study reported no difference in functional/cognitive outcome, mortality, median ICU stay, and serious adverse events between maintaining ICP at or below 20 mmHg to imaging and clinical examination alone [58, 59]. Proponents of continued ICP and CPP monitoring suggest that the study used practices that varied from established guidelines and did not specifically look into ICP monitor use for the management of intracranial hypertension, thereby limiting external validity and generalizability. ICP is an indicator of injury severity but the operational process of measuring, interpreting and making treatment decisions is complex and outcome measures such as mortality fail to address the specific contribution of ICP directed care [60]. Recent evidence looking specifically at large databases and studies following the Brain Trauma Foundation (BTF) guidelines suggest that ICP monitoring contributed to improved outcomes [61–63].
The debate regarding ICP monitoring and outcome has led investigators to seek additional, multimodal approaches to assess the physiological status of the injured brain. Multimodal monitoring includes brain oxygen monitoring (currently considered a level III clinical practice guideline recommendation) and microdialysis (not yet endorsed as a guideline). Poor short term outcome is associated with hypoxia measured by pBrO2 (partial pressure of oxygen in brain tissue) independent of elevated ICP, low CPP and injury severity [64]. The multicentered Phase II BOOST 2 (Brain Tissue Oxygen Monitoring in Traumatic Brian Injury) trial (ClinicalTrials.gov NCT00974259), estimated to complete in 2014 will evaluate whether pBrO2 levels below the critical threshold of 20 mmHg can be reduced with monitoring, in addition to the evaluation of safety, feasibility and GOSE (Glasgow Outcome Scale-Extended) scores 6 months post injury.
Microdialysis has the ability to provide information regarding the metabolic status of penumbral brain tissue, and includes real-time glucose, lactate, glycerol and glutamate measurements although robust randomized clinical trials have not yet been pursued. Studies have suggested that metabolic derangements can be detected by microdialysis prior to increases in ICP [65]. Investigators have also demonstrated that metabolic crisis, defined by brain glucose < 0.8 mmol/L and lactate/pyruvate ratio > 25 can occur at an incidence of 74 % despite adequate resuscitation and controlled ICP [66]. In rodent models, lactate levels are elevated at the site of injury after TBI [67]. Furthermore, microdialysis has been used to detect inadequate glucose levels in the brain with the use of strict systemic glycemic control [68].
While the use of pBrO2 monitoring and microdialysis has not been widely adopted in clinical use, these two devices provide investigators valuable tools beyond simple ICP measurements when evaluating emerging therapeutics. Combined microdialysis and positron emission tomography in patients following severe TBI demonstrated that metabolic crisis can even be present without cerebral ischemia as measured by oxygen extraction fraction and cerebral venous oxygen content [69].
Long Term Outcome
The long term sequelae of TBI are difficult to measure in terms of outcomes. Outcome measures of mortality and function are often used in clinical trials, but are often nonspecific in regard to the therapeutic strategy under investigation. In recent years, imaging has become an important outcome measure for TBI. Several regions of the brain are sensitive to TBI and include the hippocampus [4, 70, 71]. Long term changes in hippocampal areas such as the dentate gyrus and CA1 can stem from newborn neuron death after TBI which affects memory and causes learning deficits [72, 73]. Corpus callosum volume loss has also been demonstrated in humans and has been topographically correlated with neuropsychological outcomes (Fig. 15.2) [74]. Many studies seek to determine if early clinical intervention can translate to long term improvements. The average ICP during the acute neurointensive has been used as an early target for therapy in hopes that this indicator can correlate with long term outcome. Studies have reported that average ICP during the first 48 h do not correlate with 6 month functional nor neuropsychological outcomes [75]. However, these studies generally do not reflect continuous monitoring trends, number of spikes and waveforms, and are thus likely limited by design.
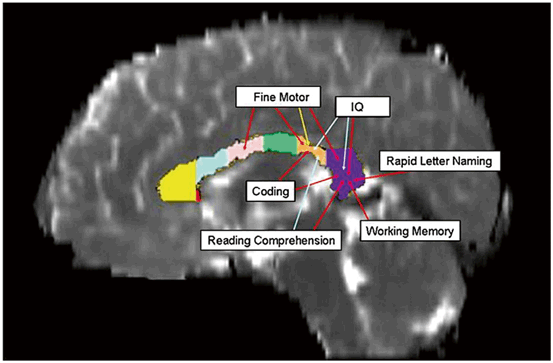
Fig. 15.2
Changes in the macro- and microstructural metrics across the callosum volume seen on diffusion tensor MRI can be topographically correlated to neuropsychological outcomes. The architecture and integrity of the high density fiber tracts of the corpus callosum are frequently altered by TBI. Fractional anisotropy (red arrows) was more frequently correlated with motor and cognitive outcomes. The mid-sagittal area (yellow arrow) of the isthmus correlated only with fine motor scores. (Reproduced from [74])
Endogenous Neurogenesis
As early as the 1990’s, investigators observed regeneration in areas of the adult mammalian brain, notably by the subependymal cells located in the subventricular zone (SVZ) following hypophysectomy [76–79]. In 2000, investigators reported the isolation of neuronal progenitor cells from human samples from the dentate gyrus [80]. In 2005, neural stem cells were isolated from the cerebral cortex of rats post TBI [81].
Proliferating and differentiating cells have been identified in the ventricular subependymal and subgranular zones of the hippocampal dentate gyrus in rat brains following TBI [82–86]. Following injury, the cells in the dentate gyrus proliferate to 3–4 fold in number, peaking at 2 days and returning to baseline at 35 days [85–87]. In rat models, TBI has been shown to promote the maturation of immature neurons. In rat models using BrDU and fluorogold tracking, cells generated in the dentate gyrus have been shown to functionally integrate via retrograde axonal tracking [88].
In mice, brain remodeling after controlled cortical injury (CCI) occurs secondary to neuronal and astrocyte proliferation and signaling activity [89]. Studies suggest that Wallerian degeneration from DAI may be sign of reorganization and healing rather than cell death [90]. NPC cells from the SVZ have been tagged and tracked as they migrate and develop into mature neurons in mice brains [91]. Stromal derived factor-1 (SDF-1), a migratory factor involved in extra-CNS homing of regenerative cells has been found to be endogenously present and required for the migration of progenitor cells from the SVZ in post TBI mice [92, 83]. In transgenic mice models, the ability to perform spatial memory tasks was eliminated by ganciclovir-mediated ablation of nestin expressing progenitor cells at time of injury [94].
Neurogenesis has also been seen outside the SVZ and hippocampus. Perilesional local neurogenesis has also been demonstrated in rats, as evidenced by the transitioning of staining from the neuroblast microtubule associated doublecortin (DCX) to the more mature NeuN neural marker [95–98]. Recently neurogenesis has been demonstrated by positive neural stem cell marker staining from samples of perilesional cortices of adult humans undergoing surgical procedures for traumatic brain injury [99, 100]. Unlike rodent models, the migration of progenitor cells from the SVZ or hippocampus to injured sites has not yet been demonstrated in humans and is likely due to the separation of the SVZ from the ependyma by a hypocellular gap [3, 101, 102]. Thus the ultimate translational relevance of the rodent data is not certain.
Therapies
The delayed nature of secondary injury allows for the possibility of potential intervention, and has been a major focus in TBI research [103]. Since the 1990’s, preclinical and clinical trials have struggled to bring novel molecular therapeutics to the bedside [104, 105]. The complexity of the post traumatic brain for targeted treatment can be demonstrated by preclinical studies of time sequence gene expression in post TBI mice, which included more than 80 genes and 24 expression sequence tags for transcription factors, signal transduction genes and inflammatory proteins [106]. Unfortunately, no specific treatment has been shown to halt or reverse neuronal death following TBI. Since the early 2000’s, investigators have explored using stem cells as potential therapy in various forms and routes of delivery.
Targets
Excitotoxicity
Glutamate has been identified as major neuroexcitatory amino acid that exacerbates cell injury, such as astrocyte swelling following traumatic brain injury [107–109]. Elevated levels can be identified in the CSF post TBI [110]. Research regarding glutamate antagonism began in the 1990’s [111–113]. Recently, the adenosine A2A receptor, found in cells such as bone marrow derived cells has been associated with increased glutamate levels following TBI. Post TBI, glutamate levels were reduced by adenosine A2A receptor inactivation or in knockout mouse models, along with reduced proinflammatory cytokines such as IL-1 and TNFα [114]. Valproate is an antiepileptic drug that has multiple targets including the GABA, sodium channel, glycogen and histone pathways, and has been demonstrated in rats to protect the BBB, reduce neural damage and improve cognition [115]. Topiramate is another antiepileptic drug that has been clinically used to reduce glutamate release after TBI in humans and a Phase II clinical trial designed to determine whether this drug can prevent epilepsy post injury is currently ongoing [116]. Investigators have even explored the benefit of caffeine and alcohol with their potential neuroexcitatory modulating mechanisms [117, 118].
Oxidative Stress
Neuroprotection with improvements in behavior has been demonstrated in rodent models for antioxidants such as deferoxamine, selenium, alpha-Phenyl-tert-N-butyl nitrone (PBN), and NXY-059 [119–122]. More recently, (-)-Epigallocatechin-3-gallate (EGCG) in green tea has been shown in post TBI rats to preserve neuronal stem cells [123]. In mice, CAPE (Caffeic phenol acid ester) improved the BBB via an antioxidant oxidant pathway [124]. Edaravone, an antioxidant and neuroprotectant scavenges NO, protects the BBB and reduced CA3 neuronal loss, apoptosis and astrocyte/glial activation in rats [125–131]. When given to a small number of TBI patients (n = 17), jugular bulb measurements demonstrated decreased reactive oxidative species [125]. Edaravone has been investigated in controlled trials for stroke but the optimal dose and therapeutic window has not yet been established [132]. Determining the optimal dosage and therapeutic window will undoubtedly be crucial in the design of a human TBI trial.
Blood Brain Barrier
The BBB has been a target of interest in TBI due to its participation in the neuroinflammatory process and the development of cerebral edema. Post injury supplementation of the endogenously expressed cyclophilin A (a protein involved in endotheial cell activation and inflammation) reduced BBB permeability 24 hr post injury in rats [133]. TIMP metallopeptidase inhibitor 3 (TIMP-3) is a MMP inhibitor that stabilizes and improves BBB integrity in animal models [134, 135]. Progesterone has been demonstrated in preclinical studies to promote endothelial progenitor cell (EPC) mediated vascular remodeling, downregulate the inflammatory cascade and decrease cerebral edema [136]. Sulforaphane (isothiocyanate) in cruciferous vegetables attenuate aquaporin-4 (AQP4) loss and improves the BBB [137]. Cannabinoid type 2 receptor agonists have been shown in mice models of TBI to improve BBB permeability and reduce macrophage/microglial activation and neuronal degeneration [138]. Citicoline, a naturally endogenous compound found to be effective in preclinical trials of BBB protection was not found to significantly improve function nor cognitive outcomes in human Phase III trials [139, 140].
Signaling Pathways
Many signaling pathways have been implicated in TBI. Erk pathway has been described as an important extracellular signal pathway in preclinical models of TBI [141]. Animal models have associated increased transcription factors such as CREB (cAMP) following TBI with changes in behavior [142]. Strategies involving histones can preserve Akt signaling, decreasing apoptosis and has been shown to increase nestin expression [143, 144]. Phosphodiesterase targeting strategies have also been explored in TBI research. PDE-4 treatment targeting the cAMP pathway improves histopathological outcomes and decreases inflammation [145, 146]. The transforming growth factor-beta (TGF-β) pathway has been implicated in mice TBI models to be linked with the Runt-related transcription factor-1 (Runx1) to promote activation and proliferation in the dentate gyrus [147]. Progesterone has been shown to regulate apoptotic protein expression in the dentage gyrus in rats but also has been to increase vasculogenesis [136, 148, 149]. Despite preclinical success, the Phase III ProTECT trial using intravenous progesterone in the acute post-TBI period was halted for futility in late 2013 (NCT00822900). The international SyNAPSe study is another Phase III progesterone clinical trial for acute TBI trial that finished enrollment in late 2013 with results now pending (NCT01143064).
Growth Factors
Growth factors are an attractive target to be either endogenously augmented or delivered exogenously to aid in the regenerative process. Intraventricular infusion of fibroblast growth factor (FGF) intro rats post TBI has been shown to increase neurogenesis in the SVZ [150]. Nerve growth factor (NGF) has been used to promote astrocyte migration and is associated with reduced apoptosis following TBI in rats. Intraventricular delivery of epidermal growth factor (EGF) has been shown to be neuroprotective in rats [151]. Vascular endothelial growth factor (VEGF) was shown to increase de novo hippocampal neurogenesis more than proliferation of resident neuroblasts in rats [152]. In mice intraventricular delivery augmented neurogenesis and angiogenesis and reduced post TBI lesion volume [153]. Gold salts have been used via local perilesion injections to reduce inflammation and apoptosis via VEGF and FGF [154, 155]. The neutrophic/mitogenic protein S100B made by astrocytes has been demonstrated to increase hippocampal neurogenesis as well as improve cognition in post TBI rats but has classically been correlated with poor outcomes when found in human CSF [156, 157]. Recently, P75, a small molecule ligand has been shown to bind neurotrophin receptors on neuronal precursors and enhance their regenerative properties in rats [158]. A randomized double blinded pilot study by Liu and colleagues is currently underway investigating repeated intranasal delivery of NGF for acute TBI (NCT01212679).
Neuronal Architecture
Cyclosporin A was shown in the 1990’s to protect axons following TBI in rats by inhibiting calcium induced mitochondrial damage [159, 160]. Preinjury inhibition of calpain, a calcium influx mediated cysteine protease preserves axonal integrity following TBI in rats [161]. Inhibitory myelin molecules such as Nogo-A has been implicated in axonal sprouting post injury, but monoclonal antibodies against this protein did not appear to act through sprouting nor cell loss protection, but still improved cognition in animal models [162]. Myelin-associated glycoprotein is another inhibitor of axonal growth, and investigators found improved sensorimotor function following intraventricular administration in rats [163]. Cyclosporin is currently being investigated in a Phase II trial.
Post-TBI Neuroinflammation
The neuroinflammatory state following traumatic brain injury is complex and likely an evolving process of dynamic cytokine expression. For example, TNF has been shown to be toxic to the neuronal stem cell proliferation phase, but not during differentiation. In fact, IFNγ enhances neuronal stem cell differentiation and neurite outgrowth [164]. This may explain why Anti-TNF or anti-IL-6 strategies have not been found to improve acute edema or motor or cognitive function in rats [165]. In preclinical studies, strategies to neutralize IL-1 have been shown to improve edema, tissue loss and cognition in mice [166, 167]. Studies have shown the local mileu in the rat brain in the first 48 h following TBI is highly proinflammatory, with elevated levels of IL-1b, IL-6, and TNFα, along with the presence of microglia and macrophages (Figs. 15.3 and 15.4) [168].
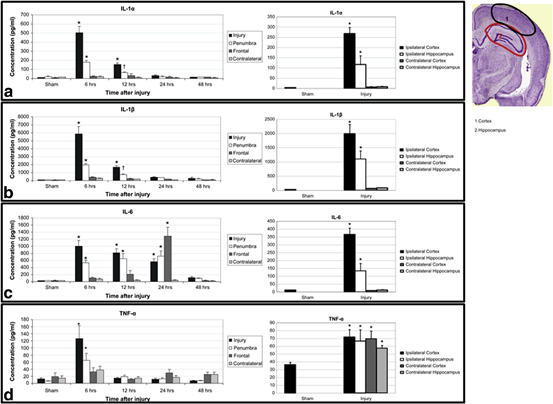
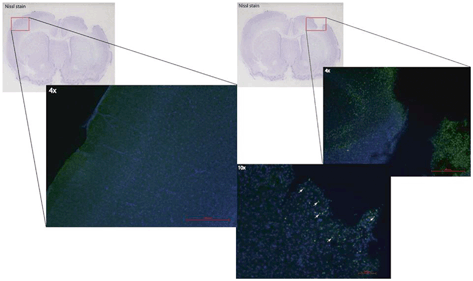
Fig. 15.3
Locoregional cytokine response following TBI in a rat model. Proinflammatory cytokines can be seen to be elevated in the brain as early as 6 h and lasting up to 24 h. IL-6 can be detected at high levels in the frontal lobes away from the impact site at 24 h. Similarly, TNF-α is elevated in a global fashion 6 h after injury compared to the local increase in the other cytokines. Note—brain section taken from Paxinos and Watson 2005. (Reproduced from Harting et al. [168])
Fig. 15.4
TBI exposes the otherwise immune privileged cortex to infiltrating macrophages and microglia that participate in the neuroinflammatory response. Brain tissue sections were isolated after TBI, and incubated anti-CD68 antibodies (green) to identify macrophages/microglia. Numerous CD68 positive cells (not seen in the contralateral hemisphere) were identified in the area of injury. (Reproduced from Harting et al. [216])
The proinflammatory environment can serve as a target of cell therapy but may limit drug efficacy or even exacerbate injury. Purely pharmacologic anti-inflammatory strategies may also interfere with complex reparative inflammatory pathways. Bortezomib a selective proteasome inhibitor used in multiple sclerosis was found to have neuroprotective properties in animal models and was associated with decreased NFkB expression [169]. TSG-6, a multifunctional immunomodulator reduces neutrophil extravasation and BBB leakage in animal models [170]. Ibuprofen has been demonstrated to improve outcome of transplanted stem cells in animal models [171, 172]. COX-2, a protein involved in the generation of inflammation mediating prostaglandins was found to be exclusively expressed in rat neurons but not astroglia and may have protective roles in only certain neurons [173, 174]. The administration of COX-2 inhibitors in a rat TBI model was found to improve cognition but worsen motor function. Thus the use of selective anti-inflammatory drugs may not be specific enough to truly target neuroprotection. Anti-inflammatory drugs such as rolipram may unfortunately increase bleeding in animal models [175]. Inhibition of single proinflammatory cascades of overlapping/redundant signal transduction pathways has not proven successful.
Neurovasculature
Investigators have also explored strategies of optimizing the neurovascular niche for resident stem cell activation and function in response to hypoxia following TBI. The hypoxia induced factor (HIF-1α) pathway has downstream components that include VEGF, SDF-1, brain derived neurotrophic factor (BDNF), tyrosine kinase receptor TrkB and associated co-receptor Nrp-1 as well as chemokine receptor CXCR4 and nitric oxide (NO) [176]. NO donor DETA/NONOate delivered via intraperitoneal injection in rats, improved proliferation, survival, and differentiation of resident neuronal stem cells [177]. Statins have also been shown to induce angiogenesis, reduce neurologic deficits, increase neuronal survival and hippocampal synaptogenesis induced angiogenesis in rats [178, 179]. In human studies, statin therapy for 10 days following moderate to severe TBI was found to reduce TNFα levels at 72 h post injury as well as disability scores at time points up to 6 months [180]. Erythropoietin is being investigated in a Phase III trial as a subcutaneous injection in patients with severe TBI under the hypothesis that secondary injury can be improved through optimizing oxygen delivery.
Biomarkers
TBI researchers have used biomarkers ranging from genes to clinical bedside measurements to evaluate injury pathophysiology as well as treatment outcomes [181, 182]. Gene expression via RNA production associated with astrocytes, phagocytes, microglia and immune-reactive cells have been described as potential biomarkers in animal models [183–188].
Biomarkers can help diagnose specific mechanisms of injury, such as BBB compromise following blast injury [189]. Elevated serum N-acetyl-asparate has been associated with DAI as well as continued secondary injury in post TBI rats [190, 191]. Microdialysis has also been used to identify axonal injury using levels of total tau and amyloid beta proteins [192, 193].
Metabolic intermediates and breakdown products have been investigated as potential biomarkers. Glycerol has been used as a marker of cell degradation and lysis [194]. Elevated plasma bilirubin, a byproduct of heme oxygenase mediated breakdown of heme and acts at free radical scavenger in mice and humans [195, 196]. Some investigators have advocated a systems biology approach for biomarker discovery, using protein network interactions as a way to screen for potential markers [197]. Common data elements have been advocated for biomarkers to improve the preclinical and clinical investigation of promising therapeutics [198, 199]. Glial to neuron ratio GFAP/ubiquitin ratio correlates to the degree of TBI on imaging such as focal versus diffuse injury patterns [200]. Biomarkers may also help identify certain cellular injury patterns that may not be identified on neuroimaging when evaluating outcomes of potential therapeutics [201].
Biomarkers have also been used to prognosticate outcomes. In human studies, increased serum IL-6, ceruloplasmin and copper levels were associated with eventual elevations in ICP following TBI [115, 202, 203]. High constant levels of CSF alpha synuculein in patients with ventriculostomies at 8 days has been associated with poor outcome [204]. Spectrin breakdown products in the CSF of patients with TBI peak at 2–3 days and indicate cytoskeletal injury and may predict injury severity and mortality [205, 206]. Studies suggest that biomarkers including serum and CSF GFAP, and CSF SBDP145 can improve the prognostic ability of scoring systems such as IMPACT [207].
Cell Therapy
In the 1990’s, Povlishock and colleagues suggested that the multiple deleterious mechanisms activated by TBI may require multiple therapies [208]. Thus single agents may be a limited and naïve strategy, even when combined. Investigators in the 2000’s began to explore the potential for stem cell therapy in neurotrauma and for CNS regeneration [209–212]. Cell therapy is an appealing therapeutic option as cells are capable of sensing and responding to environmental signals to potentially target multiple mechanisms in a sustained manner in a system where regeneration has classically thought to be limited [213]. Strategies have included both endogenous and exogenous stem cells [214]. In addition to NSC, numerous stem cell populations such as embryonic (ESC), hematopoietic (HSC) and mesenchymal (MSC) have been explored as potential cell therapy options [215–217]. In the following three decades, these cells have been applied in protocols to replace, repair, or enhance function of the post traumatic brain [218–222].
Systemic Delivery
Systemic delivery of stem cells has been explored using both intraarterial and intravenous routes. A number of stem cell types, including neuronal, mesenchymal and fetal associated cells have been utilized for systemic delivery. MSCs and NSCs have been delivered using an intraarterial strategy via the internal carotid artery for TBI in animal models but has not found popularity in human trials due to concern of ischemic embolic events (Fig. 15.5) [221, 223–225]. Intravenous delivery of cells encounter the pulmonary first pass effect described by Fischer et al (Fig. 15.6) [221, 226, 227]. The majority of mesenchymal cells are sequestered in the lung and eventually cleared by the spleen and rarely reach the brain. In fact, less than 1 % of green fluorescent protein labeled mesenchymal stem cells reached the arterial circulation. However, certain cell types have been reported to localize in the brain after intravenous delivery. Investigators using superparamagnetic iron oxide (SPIO) labeling of endothelial progenitor cells (EPC) delivered intravenously into rats have been able to detect EPCs in areas of injury on MRI and observed associated improvements in cerebral perfusion on computed tomography [228]. Nevertheless, both preclinical and clinical studies have demonstrated improvements in the brain following intravenous cell therapy suggesting a therapeutic effect that does not require direct local delivery [229, –231]. Rodent studies demonstrating BBB preservation suggests that the effect of intravenous cell therapy to the injured brain requires the participation of the spleen (Fig. 15.7) [232]. Cortical injury and increased BBB permeability is associated with a decrease in splenic mass. Following the intravenous delivery of Multipotent Adult Progenitor Cells (MAPC), CD4+ splenocyte proliferation increased, along with the production of anti-inflammatory cytokines.
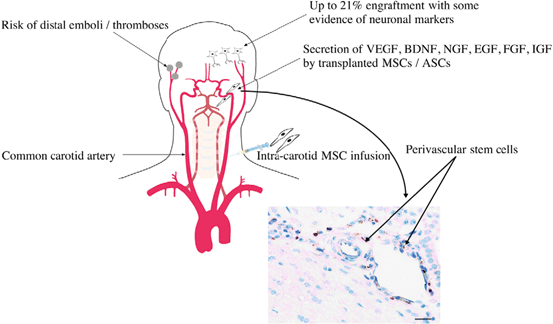
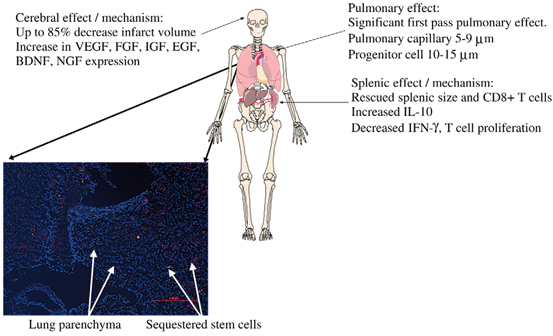
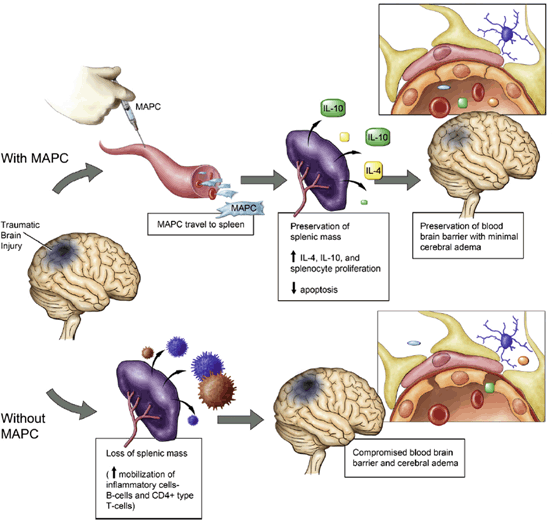
Fig. 15.5
Intraarterial introduction of stem cells. Intraarterial delivery of cells increases potential engraftment percentage due to the avoidance of the sequestration and clearance issues associated with intravenous delivery. However, the risk of distal emboli and thrombosis from intraarterial delivery can be devastating and thus this route has not been widely adopted. (Reproduced from Walker et al. [221])
Fig. 15.6
The first pass pulmonary effect plays a significant role in intravenous strategies of cell therapy. Cells labeled with quantum dots (stained red) delivered intravenously are sequestered in the pulmonary capillaries. Less than 1 % of green fluorescent protein labeled mesenchymal stem cells reached the arterial circulation. (Reproduced from Walker et al. [227])
Fig. 15.7
Proposed mechanism of Multipotent Adult Progenitor Cell mediated neurovascular protection post-TBI and the splenic interaction. TBI results in decreased splenic mass as well as increased blood brain barrier permeability. The administration of Multipotent Adult Progenitor Cells increases CD4+ splenocyte proliferation and the production of anti-inflammatory cytokines resulting in the preservation of the cerebral microvasculature and the blood brain barrier. (Reproduced from Walker et al. [232])
Fetal associated progenitor cells are also candidates for cell therapy and include umbilical cord blood and Wharton’s Jelly as well as components of the placenta. Cultured human umbilical cord blood (HUCB) cells have been demonstrated to produce cytokines and chemokines that include IL-8, MCP-1 and IL-1a. Intravenous HUCB cells reduced neurologic deficits in rats after TBI [223, 234]. Prelabeled human fetal neural progenitors have been delivered both intravenously and locally into TBI rats with the therapeutic effects likely due to angiogenesis and reduced astrogliosis rather than cell replacement [235].
The systemic delivery of stem cells has been safely used in various disease models. In preclinical and clinical trials, MSC systemic toxicity has been well studied. Adenosine A2A receptors found on bone marrow-derived cells has been implicated in mouse TBI models to involve in glutamate and inflammatory cytokine release, which was shown to lead to acute lung injury [236]. Thrombosis in stem cell transplantation has been described to cause sinusoidal obstructive syndrome of the liver (SOS) in 50–60 % of patients, with the severe form causing up to 84.3 % in mortality [237]. However, infusional toxicities associated with autologous therapy have not been demonstrated in human trials, particularly in a Phase I pediatric TBI safety trial using intravenously delivered BMMNC’s [230].
Replacement Strategies/Direct Delivery
Stereotactic transplantation of various stem cell types, including neuronal, mesenchymal, embryonic as well as induced pluripotent (IPS) stem cells into rodent brains have been shown to rescue CA3 neurons, improve cognition and neuromotor function (Fig. 15.8) [238–245]. Studies in mice where NSC injections were delivered into different ipsilateral and contralateral locations suggested that local delivery or migration of cells may not be necessary to produce functional improvements [238, 246]. In practice, the transplantation of cells directly into the injured brain has been challenging. The neuroinflammatory environment following TBI has been shown to be hostile, leading to protracted effectiveness and early loss of exogenous cells [247–250].
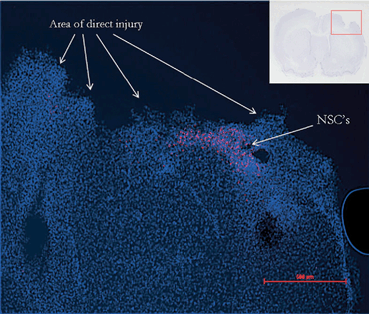
Fig. 15.8
Immunohistochemistry demonstrates the persistence of NSCs 48 h after local delivery to the penumbra of the cortical injury in a rat model of TBI. (Reproduced from [240])
Neural stem cells have even been described as biologic minipumps used for their trophic, migratory and secretory ability [251, 252]. Transplanted human NSCs have shown to produce glial derived neurotrophic factor (GDNF) resulting in axonal growth following fluid percussion TBI in rats [253]. Gene therapy such as the use of NGF transfection has also been explored to further augment the production of growth factors by modified stem cells being introduced into animal TBI models [241, 254–257].
Paracrine and Systemic Effects
MSCs have been proposed as an ideal candidate for TBI directed cell therapy due to the ability of these cells to have paracrine and systemic regenerative and anti-inflammatory effects. In vitro co-culture studies demonstrated improved NSC proliferation and expression of GFAP towards astrocyte differentiation. Human adipose tissue derived MSCs have also been demonstrated to support native NSCs in vitro [258]. Direct injection of MSCs into the injured cortex in mice promoted anti-inflammatory cytokine expression [259]. Intracranial delivery of MSCs has been applied to rodent TBI models with improved celluar as well as functional outcomes [260–263]. Contralateral cerebroventricular introduction of human umbilical cord MSCs in cyclosporine A immunosuppressed mice have been shown to increase lesional BDNF levels, decreased glial scar, improved non-phagocytic to phagocytic macrophage ratio and improved neurologic function [263]. Intrathecal MSCs have been shown in rat models to enhance the neuroprotection of NSCs via direct cell contact [264].
Embryonic stem cells (ESC) have also been used in direct transplantation. Mouse ESCs have been shown to differentiate into GABAnergic neurons as well as astrocytes [265]. However, the use of ESCs in preclinical trials has not been widely adopted due to reports that murine ESCs transplanted into rats produced tumors [266].
Direct transplantation appears to be an attractive delivery method in preclinical animal models. However, translating delivery to human applications can be challenging. The difference between the human brain post injury and animal models has been well documented. Animal heterogeneity also exists in preclinical trials [267]. Most preclinical animal models involve a unifocal injury. Human TBI injury patterns are often multifocal, and present a challenge for the local delivery of therapeutics. Green fluorescent protein labeled neural progenitor cells injected into the contralateral ventricle of mice 1 week post injury have been shown to migrate to the injured site and were detectable up to 3 months [268]. This migratory chain however, has not been demonstrated in human studies [101, 102]. The fact that TBI has multiple foci of injury if the cells don’t migrate makes direct transplantation an unattractive approach. NSCs injected locally in other human trials demonstrate little migratory capacity.
Probable Pleiotropic Mechanisms of Action of Cell Therapy
Mesenchymal Stem Cells (MSC) have been studied as a delivery mechanism for therapeutic molecules in both systemic and paracrine strategies rather than just cell replacement or transdifferentiaton [269]. MSCs have been isolated from peripheral blood, bone marrow and even adipose tissue. As an example of stem cells responding to their inflammatory environment, human bone derived MSCs cultured with extracts of TBI were induced to produce growth factors [270]. Intravenous delivery of human MSCs in rats has been shown to increase cerebral levels of NGF, BDNF, NT-3 (neurotropin-3) early after TBI and reduced apoptotic activity [271, 272]. Bone marrow derived MSCs transfected with BDNF, then given intravenously to rats showed increased BDNF in the CSF and had improved immune tolerance [273]. MAPCs used in rodent models of TBI have been shown to interact with splenocytes and T regulatory cells in the promotion of M2 regenerative microglia and the downregulation of pro-inflammatory macrophages and cytokines (Fig. 15.9) [274, 275]. The secondary injury associated with TBI leads to invariable brain volume loss over time. However, conventional MRI studies on children receiving intravenous autologous BMMNCs have demonstrated brain volume preservation up to 6 months following injury (Fig. 15.10) [230].
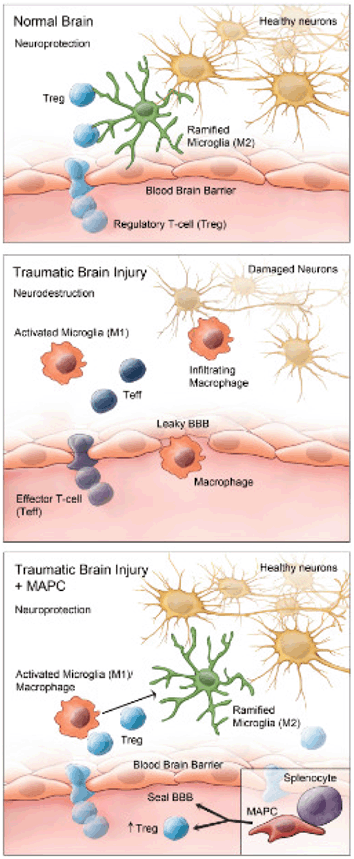
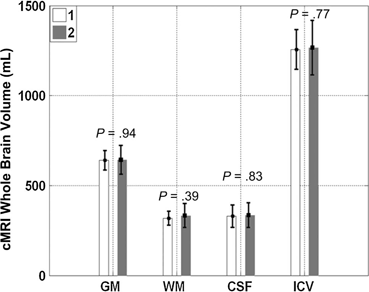
Fig. 15.9
The neuroinflammatory response following TBI involves the infiltration of neurodestructive activated M1 microglia and effector T cells across a compromised blood brain barrier. Multipotent adult progenitor cells (MAPCs), a type of MSC interacts with splenocytes, and increases anti-inflammatory cytokine production, promoting regulatory T cell activity and proliferation, which restores the blood brain barrier and shifts the neuroinflammatory state in the brain towards a M2 regenerative state. (Reproduced from [275])
Fig. 15.10
Conventional MRI (cMRI) of various brain volumes at 1 month (white bar) and 6 months (gray bar) following severe TBI in 10 pediatric patients receiving intravenous autologous bone marrow derived mononuclear cells (BMMNC). Post TBI brains are expected to undergo volume loss, but those receiving BMMNCs demonstrated preservation of brain volume over 6 months following injury. Gray matter (GM), White matter (WM), Intracranial volume (ICV). (Reproduced from [230])
Transdifferentiation
Non-neuronal progenitor cells had been proposed to act by means of transdifferentiating into neuronal cells after in vitro and in vivo studies demonstrated expression of neuronal markers. Recent studies have suggested that in vitro transdifferentiation may not actually be occurring in vivo and that the expression of neuronal markers are likely due to induced cellular stress [276]. Cells delivered intravenously rarely localize to the brain. Investigators introducing untransdifferentiated versus transdifferentiated umbilical mesenchymal cells directly into the area of injury in a rat TBI model found that the former was able to improve cognitive function and tissue morphology as well as increase neurotrophin expression [248]. Thus, the current pre-clinical strategy is to use progenitor cells, such as bone marrow derived MSCs introduced via direct or intravenous transplantation to protect native cells by creating a more favorable regenerative environment rather than inducing neuronal differentiation [277].
Other Strategies to Augment Engraftment and Function
Bioengineered constructs have been applied to help cell delivery, targeting and survival. NSCs can migrate along non-stereotypical routes for great anatomic (but short absolute distances) in immature animal models, a process that may be augmented with laminin or fibronectin based scaffolds or biodegradable nano-fibers [278, 279]. Investigators have described the concept of a biobridge that is initially comprised of transplanted stem cells that evolve to a tract with an abundance of matrix metalloproteinases, which facilitates the migration of other cells [280]. NSC induced production of connexion 43 may alter the surrounding tissue architecture [281]. NSCs interact with brain microvascular endothelial cells (BMECs) and have been used to form a model of the BBB in vitro [282]. Scaffolds can be used in vitro to model the in vivo interaction between potential cell therapies with the extracellular matrix as well as co-cultured astrocytes [283]. Scaffolds have been used to enhance the therapeutic potential of progenitor cells [227, 284–287]. FDG labeled human MSCs were placed into collagen scaffolds that are then implanted into injured rat brains. This strategy was shown to improve cell survival and energy uptake [288].
The priming and genetic enhancement of cells being used as therapeutic agents has also been investigated. GDNF enhanced neuroprogenitor cells delivered into the brain exhibit improved neuronal differentiation in rats with enhanced cognitive recovery [289]. Primed human fetal neural stem cells with agents such as progesterone improved cognition in rats when delivered post TBI [290]. Hypoxia has been shown to prime MSCs to produce growth factors such as VEGF when the cells are delivered intravenously [291, 292]. Pre-clinical animal studies have also looked at dual therapy of using intravenous G-CSF in combination with intravenously delivered human umbilical cord or bone marrow derived MSCs to maximize treatment efficacy [229, 293].
Although the intravenous route does not appear to result in the delivery of cells into the post TBI brain, strategies have been developed to increase the intracranial localization. MRI guided ultrasonic disruption of the BBB has been used as a potential strategy for the targeted local delivery of stem cells [294]. An encapsulated approach has also been used to improve biodelivery [295].
Conclusion
TBI remains challenging in both the understanding of the pathophysiology and the development of therapeutics. As the injured brain progresses from neuroinflammatory degradation to regeneration, numerous molecular and cellular processes occur that can influence outcome. While many pharmaceutical agents have been developed and tested in preclinical trials, their contribution to the complex and evolving inflammasome following TBI has not materialized in any successful clinical trials. The advancement of preclinical studies for TBI using cell therapy has been slow due to the heterogeneity of injury models, outcome measures and assays. Thus, the International Society for Cellular Therapy (ISCT) recently has recently advocated validating and strengthening standardized assays to improve the reproducibility and consistency of such data [296].
Cellular therapy offers a pharmaceutical bioreactor that can sense and interact with the inflammasome. Studies have demonstrated that inflammatory M1 to regenerative M2 phenotypic shifts can be initiated by progenitor cells. Once delivered, the fate of various stem cells needs to be verified in preclinical models. Tracking of transplanted stem cells is challenging due to issues such as auto-fluorescence and phagocytosis [250]. MicroPET and SPIO MRI can be used to monitor transplanted cells [297–300] and may be translated to humans trials [301].
Translating cell therapy from preclinical to clinical trials requires many questions to be answered. Off the shelf heterologous cells have been proposed for TBI therapy. Exogenous cells introduced either locally or systemically have not shown to engraft or survive, and thus the optimal dosing time and regimen remains unanswered. In rats, investigators have suggested that functional and behavior improvements are maximized when bone marrow derived MSCs are delivered at 7 days after injury into the corpus callosum [240, 302, 303]. Human cytokine assays demonstrate that the systemic window of therapy against pro-inflammatory cytokines can be beyond 48–72 h.
Finally, any therapy for TBI can only be deemed successful if clinical trials can demonstrate improvements in outcome. Neuroimaging has demonstrated promising volume preservation with cell therapy, but the impact of these findings to cognitive and functional outcomes are unknown and require further understanding about the role of cell therapy in the regenerative process following TBI. In the short term, a multimodal approach that includes drugs such as neurostimulants, neurorehabilitation and immunomodulatory cell therapy likely offers the best strategy to maximize recovery potential.
References
1.
Rose VL. NIH issues consensus statement on the rehabilitation of persons with traumatic brain injury. Am Fam Physician. 1999;59(4):1051–3.PubMed
2.
Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: a public health perspective. J Head Trauma Rehabil. 1999;14(6):602–15.PubMed
3.
Richardson RM, Singh A, Sun D, Fillmore HL, Dietrich DW, 3rd, Bullock MR. Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J Neurosurg. 2010;112(5):1125–38. doi:10.3171/2009.4.JNS081087.PubMed
4.
Williams S, Raghupathi R, MacKinnon MA, McIntosh TK, Saatman KE, Graham DI. In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol. 2001;102(6):581–90.PubMed
5.
Frattalone AR, Ling GS. Moderate and severe traumatic brain injury: pathophysiology and management. Neurosurg Clin N Am. 2013;24(3):309–19. doi:10.1016/j.nec.2013.03.006.PubMed
6.
Alessandri B, Nishioka T, Heimann A, Bullock RM, Kempski O. Caspase-dependent cell death involved in brain damage after acute subdural hematoma in rats. Brain Res. 2006;1111(1):196–202. doi:10.1016/j.brainres.2006.06.105.PubMed
7.
Clark RS, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci (the official journal of the Society for Neuroscience). 1997;17(23):9172–82.
8.
Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82(1):181–91.PubMed
9.
Davidsson J, Risling M. A new model to produce sagittal plane rotational induced diffuse axonal injuries. Front Neurol. 2011;2:41. doi:10.3389/fneur.2011.00041.PubMedCentralPubMed
10.
Czeiter E, Pal J, Kovesdi E, Bukovics P, Luckl J, Doczi T, Buki A. Traumatic axonal injury in the spinal cord evoked by traumatic brain injury. J Neurotrauma. 2008;25(3):205–13. doi:10.1089/neu.2007.0331.PubMed
11.
Gottesfeld Z, Moore AN, Dash PK. Acute ethanol intake attenuates inflammatory cytokines after brain injury in rats: a possible role for corticosterone. J Neurotrauma. 2002;19(3):317–26. doi:10.1089/089771502753594882.PubMed
12.
Woolf PD, Cox C, Kelly M, McDonald JV, Hamill RW. Alcohol intoxication blunts sympatho-adrenal activation following brain injury. Alcohol Clin Exp Res. 1990;14(2):205–9.PubMed
13.
Kobori N, Clifton GL, Dash PK. Enhanced catecholamine synthesis in the prefrontal cortex after traumatic brain injury: implications for prefrontal dysfunction. J Neurotrauma. 2006;23(7):1094–102. doi:10.1089/neu.2006.23.1094.PubMed
14.
Redell JB, Dash PK. Traumatic brain injury stimulates hippocampal catechol-O-methyl transferase expression in microglia. Neurosci Lett. 2007;413(1):36–41. doi:10.1016/j.neulet.2006.11.060.PubMedCentralPubMed
15.
Kobori N, Hu B, Dash PK. Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience. 2011;172:293–302. doi:10.1016/j.neuroscience.2010.10.048.PubMedCentralPubMed
16.
Zygun DA, Kortbeek JB, Fick GH, Laupland KB, Doig CJ. Non-neurologic organ dysfunction in severe traumatic brain injury. Crit Care Med. 2005;33(3):654–60.PubMed
17.
Kalsotra A, Zhao J, Anakk S, Dash PK, Strobel HW. Brain trauma leads to enhanced lung inflammation and injury: evidence for role of P4504Fs in resolution. J Cereb Blood Flow Metab (official journal of the International Society of Cerebral Blood Flow and Metabolism). 2007;27(5):963–74. doi:10.1038/sj.jcbfm.9600396.
18.
Kalsotra A, Turman CM, Dash PK, Strobel HW. Differential effects of traumatic brain injury on the cytochrome p450 system: a perspective into hepatic and renal drug metabolism. J Neurotrauma. 2003;20(12):1339–50. doi:10.1089/089771503322686139.PubMed
19.
Chu W, Li M, Li F, Hu R, Chen Z, Lin J, Feng H. Immediate splenectomy down-regulates the MAPK-NF-kappaB signaling pathway in rat brain after severe traumatic brain injury. J Trauma Acute Care Surg. 2013;74(6):1446–53. doi:10.1097/TA.0b013e31829246ad.PubMed
20.
Li M, Li F, Luo C, Shan Y, Zhang L, Qian Z, Zhu G, Lin J, Feng H. Immediate splenectomy decreases mortality and improves cognitive function of rats after severe traumatic brain injury. J Trauma. 2011;71(1):141–7. doi:10.1097/TA.0b013e3181f30fc9.PubMed
21.
Enriquez P, Bullock R. Molecular and cellular mechanisms in the pathophysiology of severe head injury. Curr Pharm Des. 2004;10(18):2131–43.PubMed
22.
Jain KK. Neuroprotection in traumatic brain injury. Drug Discov Today. 2008;13(23–24):1082–9. doi:10.1016/j.drudis.2008.09.006.PubMed
23.
Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. The J Head Trauma Rehabil. 2005;20(1):76–94.
24.
Sahuquillo J, Poca MA, Amoros S. Current aspects of pathophysiology and cell dysfunction after severe head injury. Curr Pharm Des. 2001;7(15):1475–503.PubMed
25.
Schubert A, Emory L. Cellular mechanisms of brain injury and cell death. Curr Pharm Des. 2012;18(38):6325–30.PubMed
26.
Gao X, Deng P, Xu ZC, Chen J. Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PloS one. 2011;6(9):e24566. doi:10.1371/journal.pone.0024566.PubMedCentralPubMed
27.
Giulian D, Chen J, Ingeman JE, George JK, Noponen M. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci (the official journal of the Society for Neuroscience). 1989;9(12):4416–29.PubMed
28.
Czigner A, Mihaly A, Farkas O, Buki A, Krisztin-Peva B, Dobo E, Barzo P. Kinetics of the cellular immune response following closed head injury. Acta Neurochir. 2007;149(3):281–9. doi:10.1007/s00701-006-1095-8.PubMed
29.
Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15(1):49–59.PubMed
30.
Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5(2):146–56. doi:10.1038/nrn1326.PubMed
31.
Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8(2):101–5.PubMed
32.
Walker PA, Harting MT, Baumgartner JE, Fletcher S, Strobel N, Cox CS Jr. Modern approaches to pediatric brain injury therapy. J Trauma. 2009;67(2 Suppl):S120–7. doi:10.1097/TA.0b013e3181ad323a.
33.
Bentz K, Molcanyi M, Schneider A, Riess P, Maegele M, Bosche B, Hampl JA, Hescheler J, Patz S, Schafer U. Extract derived from rat brains in the acute phase following traumatic brain injury impairs survival of undifferentiated stem cells and induces rapid differentiation of surviving cells. Cell Physiol Biochem (international journal of experimental cellular physiology, biochemistry, and pharmacology). 2010;26(6):821–30. doi:10.1159/000323991.PubMed
34.
Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab (official journal of the International Society of Cerebral Blood Flow and Metabolism). 2013b. doi:10.1038/jcbfm.2013.146.
Related posts:
 The CTX Human Neural Stem Cell Line and the PISCES Stroke Trial
Induced Pluripotent Stem Cells as a Cell-Based Therapeutic in Stroke
Issues in Clinical Trial Design in Stem Cell Trials After Stroke
The CTX Human Neural Stem Cell Line and the PISCES Stroke Trial
Induced Pluripotent Stem Cells as a Cell-Based Therapeutic in Stroke
Issues in Clinical Trial Design in Stem Cell Trials After Stroke
 Stem Cell Therapy for Neonatal Hypoxic–Ischemic Brain Injury
Stem Cell Therapy for Neonatal Hypoxic–Ischemic Brain Injury
 Biomaterials Application in Stem Cell Therapies for Stroke
Biomaterials Application in Stem Cell Therapies for Stroke
 Intra-arterial Approaches to Stem Cell Therapy for Ischemic Stroke
Intra-arterial Approaches to Stem Cell Therapy for Ischemic Stroke
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


