Perinatal Asphyxia and Trauma
John H. Menkes
Harvey B. Sarnat
The name “cerebral palsy” is thus nothing other than an invented word, the product of our nosographic classification, a label which we attach to a group of clinical cases: it should not be defined, rather it should be explained by reference to these clinical cases.
—Sigmund Freud (1, p. 3)
Although more than 160 years has elapsed since the publication of Little’s classic paper linking abnormal parturition, difficult labor, premature birth, and asphyxia neonatorum with a “spastic rigidity of the limbs” (2), the pathogenesis of cerebral birth injuries is far from completely understood. This is not because of lack of interest. The evolution and ultimate neurologic picture of cerebral palsy (i.e., the various syndromes of a persistent but not necessarily unchanging disorder of movement and posture resulting from a nonprogressive lesion of the brain acquired during development) have been recorded in innumerable papers. These include Little’s 1843 paper on spastic diplegia (2) and Cazauvielh’s 1827 monograph on congenital hemiplegia (2a), both also containing descriptions of childhood dyskinesia.
Investigations into the causes of cerebral palsy have taken various approaches. Prospective and retrospective studies have attempted to link the various neurologic abnormalities to specific disorders of gestation or the perinatal period. Pathologic studies of the brain have produced careful descriptions of various cerebral abnormalities in patients with nonprogressive neurologic disorders and have led to attempts, often highly speculative, to formulate their causes. A third line of investigation has been to induce perinatal injuries in experimental animals and to correlate the subsequent pathologic and clinical pictures with those observed in children. These approaches have been supplemented by neuroimaging studies conducted during the perinatal period and in later life. Images have been correlated with neurologic or developmental outcome or with the pathologic examination of the brain.
The various investigations have demonstrated that a given clinical neurologic deficit can be caused by a cerebral malformation of gestational origin, by destructive processes of antenatal, perinatal, or early postnatal onset affecting a previously healthy brain, or by the various processes acting in concert. Developmental anomalies are discussed in Chapter 5 and intrauterine infections in Chapter 7. This chapter considers perinatal trauma, perinatal asphyxia, and the neurologic complications of prematurity.
CRANIOCEREBRAL TRAUMA
Mechanical trauma to the central or peripheral nervous system is probably the insult that is understood best. Trauma to the fetal head can produce extracerebral lesions, notably molding of the head, caput succedaneum, subgaleal hemorrhage, and cephalhematoma.
The fetal head is often asymmetric owing to intrauterine or intravaginal pressure. The sutures override one another, the fontanelles are small or obliterated, and the tissues overlying the skull can be soft because of caput succedaneum. A caput usually appears at the vertex and is commonly accompanied by marked molding of the head. The hemorrhage and edema are situated between the skin and the aponeurosis. When the hemorrhage is beneath the aponeurosis, it is termed a subgaleal hemorrhage. As it does in a caput, blood crosses suture lines, but bleeding can continue after birth, and at times the blood loss is quite extensive.
Cephalhematoma and Subgaleal Hematoma
Cephalhematoma is a usually benign hemorrhage between the periosteum of the skull (pericranium) and the calvarium. It results from direct physical trauma or from the differential between intrauterine and extrauterine pressure. Vaginal delivery is not necessarily a prerequisite for its occurrence; it has been encountered in infants born
by cesarean section. Neonatal cephalhematoma occurs in from 1.5% to 2.5% of deliveries. Approximately 15% occur bilaterally. A linear skull fracture is seen in 5% of unilateral and in 18% of bilateral cephalhematoma (5). A depressed skull fracture may underlie a minority of cephalhematomas and cannot be detected with certainty by palpation on physical examination, so that a computed tomography (CT) scan or skull radiography may be indicated in infants with cephalhematoma and neurologic symptoms or signs. Routine ultrasound examination does not detect this lesion. Less commonly, a hematoma lies between the galea of the scalp and the periosteum. The subperiosteal hematoma is sharply delineated by the suture lines, whereas the subgaleal hematoma is not so limited and, therefore, is more diffuse. The hematoma is usually absorbed within 3 to 4 weeks, and aspiration, which can allow the introduction of infection, is contraindicated. On rare occasions, the scalp swelling is caused not by a hematoma, but by cerebrospinal fluid (CSF) that leaked from the subarachnoid compartment via a dural tear and a skull fracture. Swelling from CSF does not usually disappear in 4 weeks, and diagnosis by aspiration becomes necessary, followed by operative repair, to avoid a growing fracture(6). Although occasionally a subperiosteal hematoma calcifies (Fig. 6.1), it should cause little concern because calcium deposits are usually reabsorbed before the end of the first year, leaving no residual asymmetry.
by cesarean section. Neonatal cephalhematoma occurs in from 1.5% to 2.5% of deliveries. Approximately 15% occur bilaterally. A linear skull fracture is seen in 5% of unilateral and in 18% of bilateral cephalhematoma (5). A depressed skull fracture may underlie a minority of cephalhematomas and cannot be detected with certainty by palpation on physical examination, so that a computed tomography (CT) scan or skull radiography may be indicated in infants with cephalhematoma and neurologic symptoms or signs. Routine ultrasound examination does not detect this lesion. Less commonly, a hematoma lies between the galea of the scalp and the periosteum. The subperiosteal hematoma is sharply delineated by the suture lines, whereas the subgaleal hematoma is not so limited and, therefore, is more diffuse. The hematoma is usually absorbed within 3 to 4 weeks, and aspiration, which can allow the introduction of infection, is contraindicated. On rare occasions, the scalp swelling is caused not by a hematoma, but by cerebrospinal fluid (CSF) that leaked from the subarachnoid compartment via a dural tear and a skull fracture. Swelling from CSF does not usually disappear in 4 weeks, and diagnosis by aspiration becomes necessary, followed by operative repair, to avoid a growing fracture(6). Although occasionally a subperiosteal hematoma calcifies (Fig. 6.1), it should cause little concern because calcium deposits are usually reabsorbed before the end of the first year, leaving no residual asymmetry.
Management of a cephalhematoma is fundamentally nonoperative. Underlying skull fractures do not create a therapeutic problem and need no specific therapy unless a significant depression of bone fragments occurs.
Large cephalhematomas can result in anemia or, more often, in hyperbilirubinemia owing to absorption of hemoglobin breakdown products (7). With the advent of the vacuum extractors, there has been an increased occurrence of subgaleal hematomas. In the experience of Chadwick and his group, 89% of neonates who had experienced a subgaleal hematoma had a vacuum extractor applied to their head at some time in the course of delivery (8). Intracranial hemorrhage, skull fracture, and cerebral edema (9) can complicate a subgaleal hematoma, as can hypovolemia, coagulopathy, and jaundice, the latter consequences of extensive blood loss (8).
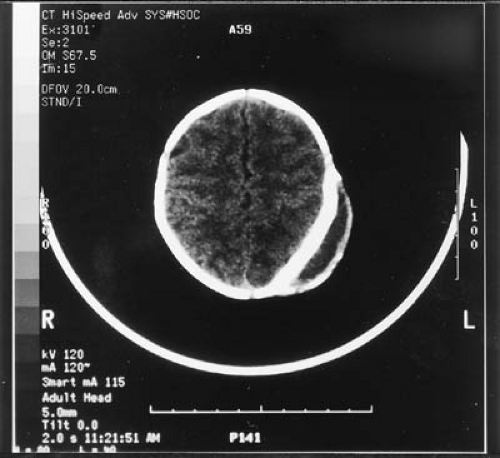 FIGURE 6.1. CT scan of a calcified cephalhematoma. (Courtesy of Dr. Franklin G. Moser, Division of Neuroradiology, Cedars-Sinai Medical Center, Los Angeles, CA.) |
Skull Fracture
The skull of the newborn is poorly mineralized and extremely pliable. These factors permit considerable distortion of the head without injury to the skull. Nevertheless, a variety of skull fractures can be seen in the newborn. These can be incurred in utero, during labor, or secondary to the application of forceps.
The most common fracture is linear and is localized to the parietal or frontal regions. When no displacement is present, the fracture should heal spontaneously, and no treatment is indicated.
A depressed skull fracture can result from pressure of the head against the pelvis. In addition, incorrect application of the obstetric forceps is often held responsible for the small, ping-pong ball depression.
Traumatic Intracranial Hemorrhage
Mechanical trauma to the infant’s brain during delivery can induce lacerations in the tentorium or cerebral falx with subsequent subdural hemorrhage. With improved obstetric techniques, large subdural hemorrhages have become relatively uncommon, generally occurring only in large full-term infants delivered through an inadequate birth canal. In the series of Gröntoft published in 1953 (10), two-thirds of infants with tentorial lacerations weighed more than 4,500 g. Similar lesions can be seen in the premature infant (11). In the more recent study of Whitby and coworkers, who subjected normal asymptomatic neonates to magnetic resonance imaging (MRI), 6.1% of infants delivered vaginally without instrumentation had subdural hemorrhages. The incidence of subdural hemorrhages was markedly increased when delivery required instrumentation. In all instances, the hemorrhage had completely resolved by 4 weeks of age, without any apparent sequelae (11a).
Small arachnoid hemorrhages are frequent with moulding of the head because of the rupture of small arachnoid bridging veins. They often are too sparce to be detected by CT, but CSF examination discloses red blood cells and increased protein. If extensive, subarachnoid hemorrhage in the neonate may provoke seizures. Only rarely are there permanent neurological sequelae to perinatal subarachnoid hemorrhage, but slowly progressive late hydrocephalus may occur in the second half of the first year.
Compression of the head along its occipitofrontal diameter, resulting in vertical molding, can occur with vertex presentations, whereas compression of the skull between the vault and the base, resulting in an anteroposterior elongation, is likely to be the outcome of face and brow presentations. Tears of the falx and tentorium can be caused by both forms of overstretch. In particular, the use of vacuum extraction can produce vertical stress on the cranium, with tentorial tears (12). Such hemorrhages are extremely common; in the series of Avrahami and colleagues published in 1993, they could be demonstrated by CT in all of 10 infants delivered by vacuum extraction (13). Most of these are minor and inconsequential. In stretch injuries, damage usually occurs where the falx joins the anterior edge of the tentorium and the hemorrhage is usually infratentorial. Tears and thromboses of the dural sinuses and of the larger cerebral veins, including the vein of Galen, are accompanied commonly by subdural hemorrhages. These can be major and potentially fatal or minor and clinically unrecognizable. The hemorrhages are mainly localized to the base of the brain; when the tears extend to involve the straight sinus and the vein of Galen, hemorrhages expand into the posterior fossa. The latter are poorly tolerated and can be rapidly fatal (14). Rarely, they can develop in utero, the consequence of motor vehicle accidents or other nonpenetrating trauma. In utero intracranial hemorrhage caused by unknown causes has been seen in infants born to Pacific Island mothers, probably the consequence of abdominal massage by traditional Pacific Island healers (15,16).
Overriding of the parietal bones occasionally produces a laceration of the superior sagittal sinus and a major fatal hemorrhage. Tearing of the superficial cerebral veins is probably relatively common. The subsequent hemorrhage results in a thin layer of blood over the cerebral convexity. Bleeding is often unilateral and usually is accompanied by a subarachnoid hemorrhage. This form of hemorrhage usually results in minimal or no clinical signs. Because the superficial cerebral veins of the premature infant are underdeveloped, this hemorrhage is limited to full-term infants (17).
Subdural hemorrhage within the posterior fossa is being increasingly recognized by neuroimaging studies. The hemorrhage can be the result of a tentorial laceration or a traumatic separation of the cartilagenous joint between the squamous and lateral portions of the occiput in the course of delivery (11). Symptoms typically appear after a lag period of 12 hours to 4 days (18). They are relatively nonspecific and differ little from those seen with intracranial hemorrhage or hypoxic-ischemic encephalopathy (HIE). They include decreased responsiveness, apnea, bradycardia, opisthotonus, and seizures (19). As the subdural hematoma enlarges, the fourth ventricle is displaced forward and soon becomes obstructed, producing signs of increased intracranial pressure. Posterior fossa hemorrhage can be accompanied by intraventricular hemorrhage (IVH) or an intracerebellar hematoma (20). An intracerebral hemorrhage is a less common result of craniocerebral trauma. It is usually seen in conjunction with a major subdural or epidural hemorrhage (4).
Gross traumatic lesions to the brainstem are uncommon. Like spinal cord injuries, they are most likely to occur in the course of breech deliveries. Injury results from traction on the fetal neck during labor or delivery, with the force of excessive flexion, hyperextension, or torsion of the spine being transmitted upward. A compression injury can ensue, with the medulla being drawn into the foramen magnum. Other instances involve laceration of the cerebellar peduncles accompanied by local brainstem hemorrhage. Generally, death occurs during the course of labor or soon after birth as a consequence of damage to the vital medullary centers (21).
Spinal cord injuries are discussed in the section dealing with perinatal injuries to the spinal cord.
HYPOXIC ISCHEMIC ENCEPHALOPATHY (HIE)
Whereas in the past mechanical damage to the brain contributed significantly to mortality during the neonatal period and to subsequent persistent neurologic deficits, mortality and neurologic deficits are now more commonly the consequences of developmental anomalies and HIE, acting singly or in concert.
HIE is the consequence of a deficit of oxygen supply to the brain. This can result from a reduced amount of oxygen in the blood (hypoxia) or a reduced supply of blood to the brain (ischemia). Hypoxia and ischemia singly or conjointly can occur during the perinatal period as a consequence of asphyxia. Many definitions exist for the term perinatal. In the context of this chapter, we restrict it to the period extending from the onset of labor to the end of the first week of postnatal life.
No generally accepted definition exists for asphyxia (22). It can be inferred on the basis of indirect clinical markers: depressed Apgar scores, cord blood acidosis, or clinical signs in the neonate caused by HIE. From the physiologic viewpoint, asphyxia is a condition in which the brain is subjected not only to hypoxia, but also to ischemia and hypercarbia, which, in turn, can lead to cerebral edema and various circulatory disturbances (4). The incidence of postasphyxial encephalopathy in Leicester, England, from 1980 to 1984 was 6 in 1,000 full-term infants, with 1 in 1,000 infants dying or experiencing severe neurologic deficits as a consequence of the asphyxial insult (23,24). More recent data compiled from Goteborg, Sweden, for the period of 1985 to 1991 showed an incidence of neonatal HIE of 1.8 per 1,000 (25).
Asphyxia can occur at one or more times during intrauterine and extrauterine life. The relative frequency of antepartum, intrapartum, and postpartum asphyxia is a
matter of considerable dispute. In the large clinical series of asphyxiated infants published by Brown and associates in 1974, the insult was believed to have occurred primarily antepartum in 51%, intrapartum in 40%, and postpartum in 9% (26). Low and coworkers, who published autopsies on perinatal deaths of full-term and premature infants in 1989, found the insult to be antepartum in 10%, antepartum and intrapartum in 40%, intrapartum in 16%, and in the neonatal period in 34% (27).
matter of considerable dispute. In the large clinical series of asphyxiated infants published by Brown and associates in 1974, the insult was believed to have occurred primarily antepartum in 51%, intrapartum in 40%, and postpartum in 9% (26). Low and coworkers, who published autopsies on perinatal deaths of full-term and premature infants in 1989, found the insult to be antepartum in 10%, antepartum and intrapartum in 40%, intrapartum in 16%, and in the neonatal period in 34% (27).
Antepartum abnormalities can either be sufficient cause for neonatal encephalopathies or risk factors that render the fetus more susceptible to asphyxia during the birth process. Volpe (4) estimated that some 70% of neonatal encephalopathy is related to intrapartum insults. Half of these infants have additional antepartum risk factors for asphyxial injury. The injury is primarily antepartum in 20% and postpartum in 10%. Based on retrospective case evaluations, Badawi and colleagues estimated that as few as 4% of term infants with neonatal encephalopathy had an insult limited to the intrapartum period without any evidence of an antepartum insult (28,29). In another 25% of infants with neonatal encephalopathy intrapartum hypoxia was superimposed on preconceptional or antepartum risk factors. These findings are in sharp contrast to those of Cowan and coworkers, who examined a stringently defined sample of neonates with neonatal encephalopathy using MRI studies and/or autopsy. Cowen and coworkers found an acute intrapartum insult in 80%, and a preexisting injury in less than 1% (30). The discrepancies between these two studies are difficult to resolve. In part they could be due to the less restricted criteria for neonatal encephalopathy used by Badawi and coworkers, who included infants with obvious chromosomal and neurodevelopmental abnormalities and those who presented with neonatal encephalopathy late in the first week of life.
A related question deals with the importance of perinatal asphyxia as a cause for cerebral palsy. Here a consensus finds that the majority of cases of cerebral palsy did not have neonatal encephalopathy. Based on retrospective MRI studies of children with cerebral palsy, Truwit and coworkers found that in 17% of patients born at term cerebral palsy was related to intrapartum asphyxia. In another 7% it was associated with intrauterine and perinatal insults (31). A Swedish population-based study compiled by Hagberg and coworkers for the period of 1991 to 1994 found that in term births intrapartum asphyxia considered severe enough to cause cerebral palsy was recorded and documented in 28% of cases. More than half of the children in this group showed extrapyramidal symptoms (31a). Using retrospective clinical analysis but excluding children with extrapyramidal cerebral palsy, Blair and Stanley estimated that in 8% of cases intrapartum asphyxia was the possible cause of brain damage (32). Nelson and Grether, who also limited themselves to children with spastic cerebral palsy, found that 6% of cases were attributable to a potentially asphyxiating complication during birth (33). In another retrospective study Gaffney and coworkers found that only 12% of all children with cerebral palsy had evidence of neonatal encephalopathy (34).
From these studies we can conclude that whereas intrapartum asphyxia is a common cause for neonatal encephalopathy, it is a not the major cause for cerebral palsy. Cerebral palsy is a heterogeneous symptom complex that in most instances is the consequence of genetic and antenatal factors and in the majority of instances is not preceded by neonatal encephalopathy (35,36,37).
Pathogenesis and Pathology
There are two facets to the pathogenesis of asphyxial brain damage: cerebrovascular physiologic factors ensuing from asphyxia, and the subsequent cascade of cellular and molecular events triggered by hypoxia-ischemia leading to cell damage and death within the central nervous system (CNS). These two aspects were reviewed by Volpe (38), Johnston and colleagues (39), and McLean and Ferriero (39a).
Cerebrovascular Physiologic Factors
Because of its relatively low energy demands, the neonatal brain has a considerable resistance to hypoxia, and most hypoxic injuries to neonates result from a combination of hypoxia and ischemia. Alterations in cerebral blood flow induced by asphyxia are therefore of primary importance in understanding the genesis of birth injuries (Fig. 6.2). Following the onset of asphyxia, cardiac output is redistributed so that a larger proportion enters the brain. This results in a 30% to 175% increase in cerebral blood flow. The increase in cerebral blood flow is induced locally by a reduction in cerebrovascular resistance and systemically by hypertension. The severity and the speed of onset of the asphyxial insult determine the cerebrovascular response (40). When asphyxia is severe and develops rapidly, cerebral blood flow decreases rather than increases, probably due to increased cerebrovascular resistance. When the hypoxic-ischemic insult is prolonged, these homeostatic mechanisms fail, cerebral vascular autoregulation is lost, cardiac output decreases, and systemic hypotension develops with reduced cerebral blood flow (41) (see Fig. 6.2).
Normal brain vasculature can compensate for the decreased cerebral perfusion by rapid dilatation of the smaller vessels, so that cerebral blood flow is maintained relatively constant as long as blood pressure is kept within the normal range. The constancy of cerebral blood flow in the face of fluctuations in systemic blood pressure is termed autoregulation. The large cerebral blood vessels are believed to be more important for cerebral autoregulation in the neonate than the arterioles, with the response to changes in blood pressure being endothelium dependent (42). A number of chemicals have been implicated in the control of cerebral arterial tone (43). Nitric oxide, by
acting on the calcium-activated potassium channel of vascular endothelium, induces vascular dilatation. Adenosine also mediates vasodilatation, whereas endothelin-1 and prostanoids mediate vasoconstriction (42,44). Hypoxia, hypercarbia, and hypoglycemia all impair cerebral autoregulation. When autoregulation becomes defective as a result of hypoxia, cerebral arterioles fail to respond to changes in perfusion pressure and carbon dioxide concentrations, resulting in a pressure-passive cerebral blood flow.
acting on the calcium-activated potassium channel of vascular endothelium, induces vascular dilatation. Adenosine also mediates vasodilatation, whereas endothelin-1 and prostanoids mediate vasoconstriction (42,44). Hypoxia, hypercarbia, and hypoglycemia all impair cerebral autoregulation. When autoregulation becomes defective as a result of hypoxia, cerebral arterioles fail to respond to changes in perfusion pressure and carbon dioxide concentrations, resulting in a pressure-passive cerebral blood flow.
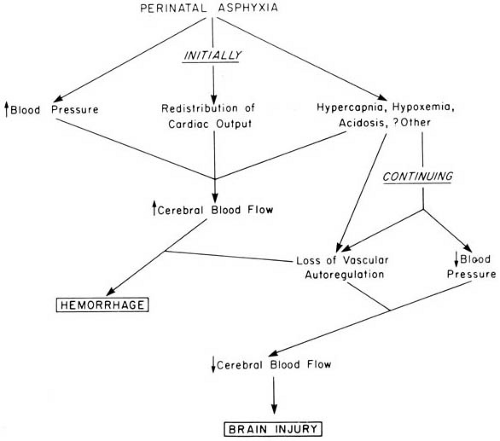 FIGURE 6.2. Interrelationships between perinatal asphyxia, alterations in cerebral blood flow, and brain damage. In addition to the mechanisms depicted, acidosis can induce focal or generalized cerebral edema, which reduces cerebral blood flow. (From Volpe JJ. Neurology of the newborn, 3rd ed. Philadelphia: Saunders, 1995. With permission.) |
It is clear that in a clinical setting multiple factors can act in concert to cause cerebral vessels to become unresponsive to systemic blood pressure (45,46). Although in the preterm infant the lower limits of autoregulation are very close to the mean systemic arterial pressure, adequate cerebral perfusion can be maintained as long as the mean arterial blood pressure ranges between 24 and 39 mm Hg (47). When hypotension exceeds these lower limits, the preterm infant is unable to compensate for the drop in blood pressure. With the arteriolar system unable to respond to decreased perfusion pressure by vasodilatation, there is a striking reduction in cerebral blood flow (47).
After termination of the ischemic insult there is a marked increase in cerebral blood flow, probably the result of the various vasodilating factors already cited. This early increase in cerebral perfusion is followed by a decline and a second, delayed increase in cerebral blood flow, probably the consequence of an increased synthesis of nitric oxide. It is during this second phase that most of the deleterious events occur that lead to cell death within the brain.
Asphyxial brain injury is similar regardless of whether the brain has incurred a global asphyxial insult as occurs in perinatal asphyxia, hypoperfusion as after cardiac arrest (see Chapter 17), or focal ischemia as after a vascular occlusion (see Chapter 13). Some authors (and lawyers) attribute great significance to meconium as a cause of intrapartum or neonatal asphyxia, but the expulsion of meconium into the amniotic fluid in the first place often is due to a generalized parasympathetic discharge with increased peristalsis that results from fetal distress or an hypoxic-ischemic insult in utero. Aspirated meconium at delivery can complicate the problem further by obstructing the infant’s airway, but meconium per se is not a cause of asphyxia.
The mechanisms for brain damage in asphyxial brain injury are not completely clear. Volpe, in reviewing the physiologic aspects of asphyxial injury, suggested that the loss of vascular autoregulation coupled with hypotension reduces cerebral blood flow to the point of producing tissue necrosis and subsequent cerebral edema (4). Combined clinical and imaging studies by Lupton and associates in which intracranial pressure of asphyxiated term infants was correlated with their CT scan corroborate Volpe’s view that tissue necrosis precedes cerebral edema rather than vice versa, with maximum abnormalities being seen between 36 and 72 hours after the insult (48). Nevertheless, it
is still likely that tissue swelling can to some extent further restrict cerebral blood flow and cause secondary edema. The earliest phase of cerebral edema probably reflects a cytotoxic component, whereas a vasogenic component characterizes the edema that accompanies extensive tissue injury (49). In asphyxiated newborns, increased intracranial pressure after perinatal asphyxia is a relatively uncommon complication; in the series of Lupton and coworkers it was encountered in only 22% of asphyxiated infants (48). It is important that the clinician recognize that a bulging fontanelle and split sutures in the neonate after an asphyxiating or severe ischemic insult does not signify potentially reversible cerebral edema after obstructive hydrocephalus, mass lesions such as subdural hematomas, and meningitis have been excluded, but rather irreversible encephalomalacia with massive necrosis in both gray and white matter, and hence is a very bad prognostic sign.
is still likely that tissue swelling can to some extent further restrict cerebral blood flow and cause secondary edema. The earliest phase of cerebral edema probably reflects a cytotoxic component, whereas a vasogenic component characterizes the edema that accompanies extensive tissue injury (49). In asphyxiated newborns, increased intracranial pressure after perinatal asphyxia is a relatively uncommon complication; in the series of Lupton and coworkers it was encountered in only 22% of asphyxiated infants (48). It is important that the clinician recognize that a bulging fontanelle and split sutures in the neonate after an asphyxiating or severe ischemic insult does not signify potentially reversible cerebral edema after obstructive hydrocephalus, mass lesions such as subdural hematomas, and meningitis have been excluded, but rather irreversible encephalomalacia with massive necrosis in both gray and white matter, and hence is a very bad prognostic sign.
Cellular and Molecular Events Triggered by Hypoxia-Ischemia
Over the last few years increasing attention has been paid to the molecular and cellular aspects of cell death within the nervous system. Gluckman et al. distinguished two phases (50). The first phase occurs during the insult and the immediate period of reperfusion and reoxygenation. The second phase evolves after a period of some hours and extends for at least 72 hours. During the first phase, asphyxia rapidly results in the conversion of nicotinamide adenine dinucleotide (NAD) to reduced NADH. As the energy demands fail to be met, there is a shift from aerobic to anaerobic metabolism, causing acceleration of glycolysis and increased lactate production. In experimental animals, brain lactate increases within 3 minutes of induction of asphyxia (51). At the same time, the concentration of tricarboxylic acid cycle intermediates decreases and the production of high-energy phosphates diminishes. These changes result in a rapid fall in phosphocreatine and a slower reduction in brain adenosine triphosphate (ATP) concentrations. With reduction of ATP levels the various ion pumps, notably the Na+–K+ pump, the most important transporter for maintaining high intracellular concentrations of potassium and low intracellular concentrations of sodium, becomes inoperative. As ion pump function is lost, the neuronal membrane begins to change. Some neurons, such as the CA1 and the CA3 hippocampal neurons, hyperpolarize, whereas others, such as the dentate granule cells, depolarize. If anoxia persists, all cells undergo a rapid and marked depolarization with complete loss of membrane potential.
The aforementioned changes in lactate and high-energy phosphates can be documented in the asphyxiated infant by proton and 31P magnetic resonance spectroscopy. These studies show an early increase in lactate (52). A decrease in phosphocreatine during the initial insult is reversed on resuscitation but is followed by a slow, secondary decline some 24 hours later. Intracellular pH and other indices of cellular energy status frequently remain normal for the first day of life (53,54).
Decreases in intracellular and extracellular pH precede changes in membrane potential as hypoxia induces production of lactate and intracellular acidosis. The decrease in extracellular pH is believed to be the consequence of extrusion of intracellular hydrogen ions, intracellular lactate, or both. At the time when the neuronal membrane potential is abolished, there are a number of striking ionic changes. They include an efflux of potassium and an influx into cells of sodium, chloride, and calcium. The increase in intracellular calcium appears to play a critical role in cellular injury. It results from a failure of energy-dependent calcium pumping mechanisms and an opening of voltage-dependent calcium channels. The accumulation of intracellular calcium in turn initiates a cascade of deleterious events:
Activation of phospholipases. These induce membrane injury and the release of arachidonic acid, which in turn produces large amounts of oxygen radicals.
Activation of proteases. These disrupt the microtubules and cytoskeleton.
Activation of nucleases. These cause an injury to the nucleus.
Activation of nitric oxide synthetase. This results in an overproduction of nitric oxide, which has neuronal toxicity on its own or when converted to peroxynitrite. One target of nitric oxide and peroxynitrite is mitochondria, and with severe hypoxic-ischemic insults there can be complete mitochondrial failure.
Glutamate release. Among the numerous factors responsible for asphyxial neuronal damage, glutamate release and the resultant excitotoxicity have received the most attention and are probably the most important determinants for neuronal death. Increased glutamate is the consequence of increased release of the neurotransmitter and impaired reuptake (55). The mechanism for increased glutamate release is controversial, and there is evidence that it may not be entirely calcium dependent, but may also be due to a reversal of the neuronal glutamate transporters, which instead of removing extracellular glutamate, release glutamate (56). Whatever the mechanism, the result is an excessive stimulation of the neuronal excitatory receptors. This is evidenced by focal elevations in the regional cerebral glucose metabolism in basal ganglia and cerebral cortex in infants who sustained asphyxial brain damage (57).
Glutamate also binds to postsynaptically located glutamate receptors that regulate calcium channels, and its release results in a further increase in intracellular calcium. Several other mechanisms also have been implicated in the increase of excitatory amino acids. These reactions were reviewed by Berger and coworkers (58) and Johnston and coworkers (39,59).
How excitotoxins induce cell death is not completely clear. Rothman and Olney proposed that prolonged neuronal depolarization induces both a rapid and a slowly evolving cell death (60). Rapid cell death is caused by an excessive influx of sodium through glutamate-gated ion channels. This leads to the entry of chloride into neurons. The increased intracellular chloride induces further cation influx to maintain electroneutrality, and the chloride and cation entry draws water into cells with ultimate osmotic lysis. Most important, calcium influx occurs. A sustained increase in intracellular calcium induces the “toxic cascade,” outlined above whose end result is cell death by necrosis (61,62).
Release of neuron-specific enolase (NSE). NSE is a glycolytic enzyme present in all classes of neurons, and concentrations in blood and CSF of normal infants and children can be measured and are reliable (62a). NSE becomes markedly elevated in the serum and CSF following cardiac arrest, stroke, or other lesions that cause necrosis of neurons (62b). In neonatal rats, NSE elevation after hypoxia is attenuated by lactate (62c), and in adult mice, Bcl-2 is under the control of NSE and is becomes overexpressed in hippocampal CA1 and dentate gyrus neurons after hypoxic-ischemic insults; the importance of this is that Bcl-2 suppresses apoptosis and delays cell death, and hence may play a protective role (62d). NSE levels in CSF also may provide a useful clinical marker of cerebral infarction or other irreversible brain damage in neonates. NSE immunoreactivity can be readily detected in neurons in tissue sections and has been used for years by pathologists as a neuronal marker.
Inflammatory reactions involving a variety of cytokines may also contribute to hypoxic-ischemic cell death. The increase in cytokines could stem from an infection, notably a chorioamnionitis that predates the hypoxic-ischemic insult, or it could result from the activation of microglia by asphyxia (63,64).
The late phase of asphyxial injury and cell death is marked by an inappropriate induction of apoptosis. Apoptosis refers to the activation of genetically determined cell-suicide programs. Choi proposed that a single insult might trigger both excitotoxic necrosis and apoptosis, with the severity and duration of the insult determining which death pathway predominates (61). In regions containing large amounts of apoptosis inhibitors, necrosis predominates; conversely, in the absence of endogenous apoptosis inhibitors, hypoxia-ischemia induces apoptosis (39a). Nakajima and coworkers found that apoptosis is more prevalent than necrosis in the hypoxic-ischemic newborn brain (65), suggesting that the impact of apoptosis-executing caspases, key effectors of apoptotic death, is much greater in the immature than in the mature brain (66). The factors that promote postasphyxial apoptosis are under intense investigation. They are believed to include free radicals, increased expression and enhanced concentrations of inflammatory cytokines, and alterations in the concentrations or the response to endogenous growth factors (67). The observation that neurotropins, such as brain-derived neurotrophic factor, act as neuroprotectors after a hypoxic insult provides evidence for the importance of neurotropins in mediating postasphyxial brain injury (68). Asphyxia also induces both rapid and delayed changes in the transcription of several genes, notably c-fos, c-jun, and some of the heat-shock proteins (69, 70,71). These substances are believed to have a significant influence on the extent of apoptosis (70). The contributions of hypoglycemia and intracellular acidosis, whether caused by accumulation of lactic acid or products of ATP hydrolysis, to the extent and severity of asphyxial brain damage have not been resolved (72).
A large number of strategies aimed at blocking the postasphyxial events that lead to neuronal damage have been proposed. These involve inhibition of glutamate release, blockade of glutamate receptors, inhibition of the cytokine effects, and blockade of apoptotic cell death. None has had any significant clinical application (39,73,74). They are reviewed by Berger and coworkers (58). More recently, the effectiveness of growth factors such as erythropoietin and brain-derived neurotrophic factor in improving outcome after an asphyxial insult has been encouraging (39a).
Imaging of the Neonatal Brain after Hypoxic-Ischemic Encephalopathy
The findings in imaging studies of the neonatal brain after hypoxic-ischemic injury depend on several factors, most of them temporal: the gestational age of the infant; the time since the insult; and whether there was a single temporal event or repeated or chronic hypoxic or ischemic periods. Ultrasound studies are the most accessible because they can be performed noninvasively at the bedside in even the sickest infants, through the anterior fontanelle, and can be compared with prenatal ultrasound studies of the fetal head and also performed serially postnatally. They are excellent for demonstrating ventricular size, and often can detect cerebral edema, periventricular lesions and hemorrhages and infarcts. Ultrasound is not very precise for structures far from the midline, such as the cerebral cortex, nor for posterior fossa structures.
Computed tomography offers better resolution than ultrasound and is very good for detecting intracerebral hemorrhages, calcifications, ventricular size, and periventricular leukomalacia, but it lacks the detail provided by MRI as well as the versatility of different MRI modalities. CT can be performed easily in neonates, even those on ventilators, and does not require anesthesia, but the infant must be transported to the radiology suite.
Magnetic resonance imaging provides the most information but has the limitation of not being accessible to very sick infants on life support who cannot be moved and are connected to a ventilator. In chronic states, when the infant is more stable, three distinctive patterns are demonstrated by MRI following hypoxic-ischemic injuries: periventricular leukomalacia occurs mainly in preterm infants with subacute or chronic hypoxia-ischemia; basal ganglionic and thalamic infarcts occur in term neonates with profound asphyxia; multicystic encephalomalacia occurs in a minority of infants with severe encephalopathy following relatively mild hypoxic-ischemic events (74a). Diffusion-weighted MRI of the neonate can identify early injury after an insult because of its ability to detect subtle alterations in brain water content (39a,74b–74c). Magnetic resonance spectroscopy (MRS), especially if done as a three-dimensional study in the neonatal brain, can detect metabolites such as lactate, N-acetyl aspartate, choline, and creatine that provide functional data regarding metabolic integrity in specific regions of the brain (74c,74e).
Selective Vulnerability
Whatever the biochemical and physiologic mechanisms for brain damage, the relative resistance of the neonate’s brain to hypoxia has been known for some time. Probably this phenomenon reflects a slower overall cerebral metabolism and smaller energy demands by the brain of the neonate compared with that of the adult. Total metabolism of the brain of a newborn mouse is approximately 10% that of the adult mouse’s brain, and glycolysis also proceeds at a much slower rate (75). In that respect the relative resistance of the cardiovascular system to hypoxic injury also can be operative.
The factors that determine the selective vulnerability of certain neuronal populations are incompletely understood. In part, regional distribution of injury reflects the vascular supply to the brain, with the injury being maximal in the border zones between the major cerebral arteries. In the striatum, the topography of neuronal death is probably related to the density of excitatory receptors and the expression of the various receptor subtypes (76). The Myers model of “partial” versus “total” asphyxia, resulting in different sites of the principal lesions in neonatal monkeys, cannot be extrapolated to the human condition. This is because the monkey brain is more mature at birth, in terms not only of structure, synaptic organization, and myelination, but also in terms of better autoregulation of cerebral blood flow. In addition, the monkey brain is considerably smaller than the human brain, with a shorter distance for blood flow to reach terminal perfusion, and blood vessels are narrower than in the human neonate. Human cerebral arterioles do not acquire their muscular walls until near term, an anatomic prerequisite for autoregulatory function in cerebral blood flow, whereas the monkey already has mature cerebral vasculature for several weeks before birth.
It is this combination of vascular and metabolic factors that results in the various distinct pathologic lesions that have been well described by classic pathologists over the course of the last century.
Multicystic Encephalomalacia
The neonatal brain responds to infarction differently than the mature brain. Rather than dense gliotic scars, pseudocysts are the usual long-term residual lesions. The reasons for the formation of cysts in the newborn brain are that areas of infarction tend to be relatively larger than in the adult because collateral circulation is less well developed and because the ability of neonatal brain to mobilize reactive gliosis is limited. The number of astrocytes per volume of neonatal brain is approximately one-sixth that of the adult brain in both gray and white matter; hence the response to injury is not nearly as effective and the glial cells present are only able to form thin septa without neurons, compartmentalizing the empty space after macrophages have cleared away the necrotic tissue. These glial septa create the multiple pseudocysts of multicystic encephalomalacia. They are pseudocysts rather than true cysts because they are not lined by an epithelium, as are ependymal cysts. Multicystic encephalomalacia is therefore the end result of extensive cerebral infarcts.
When the primate fetus is subjected to acute total asphyxia, a reproducible pattern of brain disorders ensues (77,78). This pattern includes bilaterally symmetric lesions in the thalamus and in a number of brainstem nuclei, notably the nuclei of the inferior colliculi, superior olive, and lateral lemniscus. The neurons of the cerebral cortex, particularly the hippocampus, are especially vulnerable, as are the Purkinje cells of the cerebellum (4,79).
Soon after the initial insult, the first changes observed using electron microscopy are in the neuronal mitochondria, the internal structure of which becomes swollen and disrupted (80). Gradual widespread transneuronal degeneration follows. With progressively longer periods of total asphyxia, the destructive changes in the thalamus become more extensive, and damage begins to appear in the putamen and in the deeper layers of the cortex. (Fig. 6.3). In its extreme form, asphyxiated animals show an extensive cystic degeneration of both cortex and white matter. Connective tissue replaces the damaged areas in the forebrain, but a relative lack of cellular reaction occurs in the central nuclear areas (78).
This experimentally produced picture resembles cystic encephalomalacia (central porencephaly, cystic degeneration) of the human brain, a condition characterized by the formation of cystic cavities in white matter (Fig. 6.4). When small, the cysts are trabeculated and do not communicate with the ventricular system. In their most extensive
form they can involve both hemispheres, leaving only small remnants of cortical tissue. The cavities are generally believed to be the products of insufficient glial reaction, perhaps the result of cerebral immaturity, or to reflect the sudden and massive tissue damage caused by the aforementioned circulatory or anoxic events. This pathologic picture is seen not only as a consequence of severe perinatal asphyxia, as first established by Little (2), but also in twin pregnancies after intrauterine fetal death and in fetal viral encephalitides such as herpes simplex (81,82,83). Infants surviving this type of insult usually develop a severe form of spastic quadriparesis.
form they can involve both hemispheres, leaving only small remnants of cortical tissue. The cavities are generally believed to be the products of insufficient glial reaction, perhaps the result of cerebral immaturity, or to reflect the sudden and massive tissue damage caused by the aforementioned circulatory or anoxic events. This pathologic picture is seen not only as a consequence of severe perinatal asphyxia, as first established by Little (2), but also in twin pregnancies after intrauterine fetal death and in fetal viral encephalitides such as herpes simplex (81,82,83). Infants surviving this type of insult usually develop a severe form of spastic quadriparesis.
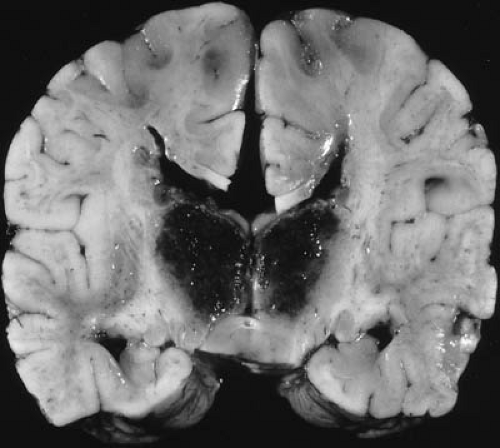 FIGURE 6.3. Coronal section of cerebral hemispheres at autopsy of a 9-month-old boy with bilateral thalamic hemorrhages. This is part of the pattern of deep infarction due to severe and prolonged perfusion failure of the brain secondary to shock. Hemorrhagic infarcts are more common than ischemic infarcts in the thalamus of fetuses and young infants. The hemorrhages do not extend into the lateral or third ventricles, and the cerebrum is otherwise normally developed and without other lesions, though microscopically it showed extensive neuronal changes of hypoxic-ischemic encephalopathy (not shown). |
The pathologic differentiation between cystic degeneration and hydranencephaly is discussed in Chapter 5.
Selective Neuronal Necrosis and Laminar Necrosis of the Cortex
The distribution of cerebral lesions induced by acute total asphyxia rarely reproduces the distribution of lesions found in infants who have survived partial but prolonged asphyxia. When prolonged partial asphyxia is induced experimentally, primates develop high carbon dioxide partial pressure (pCO2) levels and mixed metabolic and respiratory acidosis (78,84). These are usually accompanied by marked brain swelling, which compresses the small blood vessels of the cerebral parenchyma. The resultant increase in vascular resistance superimposed on the systemic alterations leads to various focal cerebral circulatory lesions whose location is governed in part by vascular patterns and in part by the gestational age of the fetus at the time of the asphyxial insult (85,86). Selective necrosis of neurons may be followed by mineralization of those cells.
The neonatal cerebral cortex is vulnerable to laminar necrosis after a severe ischemic insult. This selective neuronal necrosis involves some layers more than others. In preterm and term infants, layers 3 and 5, which contain pyramidal cells, are most vulnerable, but in later infancy and childhood, layer 4 is most severely affected. This layer contains granule cells that are sensory, rather than motor, in function. Layer 4 is largest in the striate (occipital) cortex, where it is the principal visual receptive zone. Laminar necrosis is extensive degeneration of neurons in the affected layers, with relatively better preservation in other layers, though pyknosis and karyorrhexis indicating dying neurons are seen in neurons in all layers. These changes may be expressed in infants who survive as cortical blindness and spastic diplegia, though damage in other parts of the brain, such as the lateral geniculate body and periventricular leukomalacia also contribute to the clinical deficits. Laminar necrosis may be identified in MRI, particularly in fluid-attenuated inversion recovery (FLAIR) sequences, as a bright line of increased signal within the cortex and parallel to the surface of the brain (Fig. 6.5).
Periventricular Leukomalacia
One lesion that occurs with particular frequency in the premature infant is periventricular leukomalacia (PVL) (Figs. 6.6 and 6.7). First delineated by Banker and Larroche (87), this condition consists of a bilateral, fairly symmetric necrosis having a periventricular distribution. The two most common sites are at the level of the occipital radiation and in the white matter around the foramen of Monro (88,89). In addition, there can be diffuse cerebral white matter necrosis that usually spares the gyral cores (90). Preterm infants of 22 to 30 weeks’ gestation tend to experience more widespread and confluent periventricular necrosis, whereas older premature infants exhibit more-focal necrosis (91).
The evolution of PVL has been studied by neuropathologic and neuroimaging methods. Within 6 to 12 hours of the suspected insult, coagulation necrosis occurs in the affected areas, accompanied by proliferation of astrocytes and microglia, loss of ependyma, and, in some cases, subcortical degeneration. Focal axonal disruption and death of oligodendroglia are some of the earliest signs of injury, with the developing oligodendroglia being especially vulnerable (4).
The pathogenesis of PVL is uncertain and is most likely to be multifactorial. Five major factors are believed to be operative.
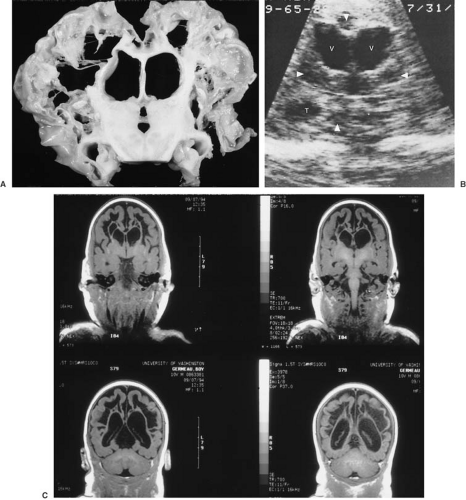 FIGURE 6.4. Cystic encephalomalacia. A: Coronal section of cerebral hemispheres of a 14-month-old boy with multicystic encephalomalacia. This is the gross pathologic appearance of lesions similar to those seen in panel C by MRI. The architecture of the cerebral white matter and cerebral cortex, including the gyri, is severely disrupted by these extensive infarcts and cerebral atrophy. The lateral and third ventricles are dilated to compensate for the atrophy. Deep structures appear better preserved, but microscopically also showed extensive neuronal loss and microinfarcts. B: Coronal sonogram of the same infant. Moderate ventriculomegaly and numerous poorly defined anechoic areas in periventricular parenchyma and in basal ganglia are visible (arrowheads) (V, lateral ventricles; T, temporal horn of lateral ventricle). Ultrasonography was performed at 7 months of age, autopsy at 16 months. The infant had a history of seizures and profound developmental retardation. In this instance, the most likely cause for the condition appeared to have been a cytomegalovirus infection. C: Coronal T1-weighted MRI of a 9-week-old boy, born at 31 weeks’ gestation, showing multicystic encephalomalacia throughout the white matter. The ventricles are large because of the atrophy of cerebral parenchyma, both gray and white matter. This is the result of severe hypoxic/ischemic encephalopathy in the perinatal period. (Panel B, from Stannard MW, Jimenez JF. Sonographic recognition of multiple cystic encephalomalacia. Am J Neuroradiol 1983;4:11. With permission.) |
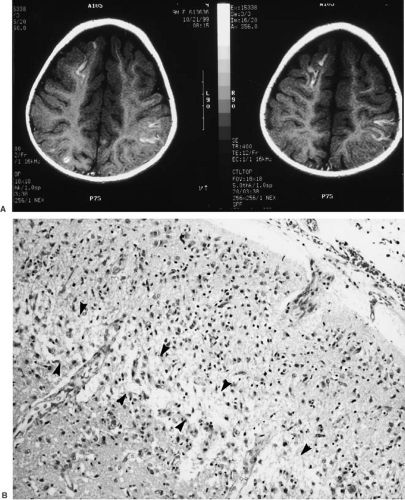 FIGURE 6.5. A: MRI-T2 fluid-attenuated inversion recovery (FLAIR) sequence of a term neonate with laminar necrosis of the cerebral cortex. The bright signal within the cortex corresponds to necrosis in layer 4. B: Section of occipital lobe of a 5-year-old girl who suffered severe perinatal asphyxia and also had congenital hydrocephalus, successfully shunted in the neonatal period. She had an additional hypoxic event 3 weeks prior to death. Dark, shrunken nuclei of dying and dead neurons are seen in all layers (arrows), but layer 4 is undergoing actual necrosis of the entire layer, known as laminar necrosis of the cortex. |
A failure in perfusion of the periventricular region. The distribution of the focal necrotic changes of PVL suggests inadequate circulatory perfusion and infarction in the arterial end zones, areas that are most susceptible to a fall in cerebral blood flow and reduced perfusion (92). The anatomic picture indicating a low blood flow to cerebral white matter has been confirmed by cerebral blood studies (93).
A second factor in the pathogenesis of PVL is derived from the observation by Doppler ultrasound studies that cerebral vascular autoregulation is impaired in a substantial proportion of premature infants, with a
propensity for pressure-passive circulation (94). Loss of autoregulation is particularly common in preterm infants who have experienced hypoxic-ischemic events (94). Because even in healthy preterm infants white matter has an extremely low perfusion, the vulnerability of the periventricular region to ischemia becomes readily explicable (45,93). Experimental work has demonstrated that hypotension induced by exsanguination or by administration of endotoxin results in reduced perfusion of periventricular white matter and occipital white matter. By contrast, these measures do not induce any significant reduction in blood flow to the cerebral cortex or to the deep gray matter nuclei (95). In substantiation of the clinical importance of impaired autoregulation in the induction of PVL, Volpe demonstrated that the subset of premature infants with pressure-passive cerebral circulation have an extremely high incidence of PVL (96).
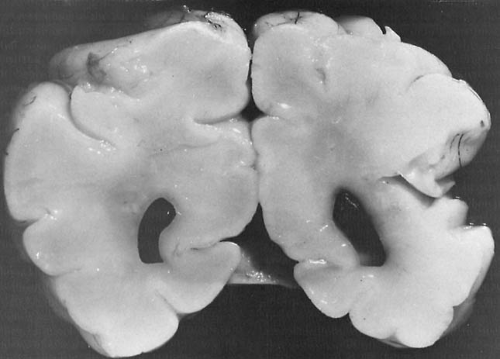
FIGURE 6.6. Periventricular leukomalacia. Semicircular areas of malacia surround both lateral ventricles. (From Cooke RE. The biologic basis of pediatric practice. New York: McGraw-Hill, 1968. With permission.)
A third factor in the pathogenesis of PVL pertains to the intrinsic vulnerability of the early-differentiating oligodendroglia (preoligodendroglia, i.e., cells at a developmental stage before the acquisition of myelin) to excitatory neurotransmitters, such as glutamate, and to attack by free radicals (97,98,99). This vulnerability may be the consequence of a lack of such antioxidant enzymes as catalase and glutathione peroxidase during a period when oligodendroglia undergo rapid iron acquisition (96,100). Elevated levels of lipid peroxidation products have been found in the CSF of premature infants who had evidence of white matter injury by MRI (99).
An increasing amount of clinical and experimental evidence shows that cytokines play an important role in the induction of white matter damage (98,101). The administration of interferon-alpha 2a to term infants as treatment for hemangiomas has resulted in spastic diplegia and delayed myelination. In some instances, diplegia did not resolve with discontinuation of cytokine therapy (102). Retrospective assays of neonatal blood have shown that preterm and term children with spastic diplegia had higher blood levels of various cytokines, including interferon-alpha, interferon-gamma, interleukin 6 (IL-6), IL-8, and tumor necrosis factor (TNF)-alpha, than did control children (103) It is of note that the association between elevated cord blood levels of cytokines and the development of white matter damage is weaker in premature than in term infants (104). In the study of Grether and colleagues, serum interferon levels were elevated in 78% of children with spastic diplegia but only in 20% of children with hemiparesis and in 42% of children who developed quadriparesis (103). Cytokines can also be demonstrated
by in situ immunohistochemical methods in neurons in the neocortex, hippocampus, basal ganglia, and thalamus of infants with PVL (105). It appears likely that cytokines such as interferon-alpha, interferon-gamma, tumor necrosis factor-alpha, IL-6, or IL-8 might damage white matter by leading to hypotension or by inducing ischemia through intravascular coagulation. Cytokines also could have a direct adverse effect on developing oligodendroglia or induce the product of other cytokines such as platelet-activating factor, which can further damage cells (106).
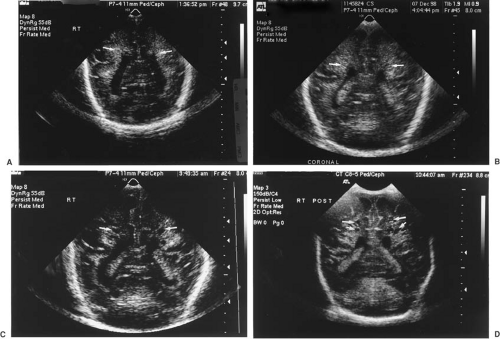
FIGURE 6.7. Evolution of cystic periventricular encephalomalacia. Ultrasound, coronal views. A: At 4 days of age, there are focal echodense areas bilaterally in the periventricular white matter (arrows). B: At 9 days of age, the bilateral periventricular echogenicity is more clearly evident (arrows). C: At 23 days of age, early cystic changes are seen bilaterally in the periventricular region (arrows). These are more severe on the right. D: At 1 month of age, multiple periventricular cystic changes are seen (arrows). This boy was the 1,445-g product of a 30-week twin pregnancy. His neonatal course was complicated by recurrent apnea and bradycardia. A septic work-up was negative. Neurologic examination was unremarkable but for jerky movements of the extremities. At 5 months of age, this youngster had spastic diplegia most severe in the trunk and lower extremities. (Courtesy of Dr. Nancy Niparko, Cedars-Sinai Medical Center, Los Angeles, CA.)
The role of hypocarbia during the first days of life in mechanically ventilated premature infants in predisposing them to PVL has been suggested by several studies (107). Fritz and coworkers proposed that hypocarbia reduces cerebral blood flow and decreases tissue oxidative metabolism, with increased intracellular calcium and the various secondary events already described (108).
From a clinical point of view, spastic diplegia is the most common and most consistent sequela of PVL. It is nearly always bilateral, although often asymmetric in severity. Because of the propensity of the periventricular necrotizing lesions to appear earliest and most prominently around the occipital horns of the lateral ventricles, optic radiation fibers may be involved and sometimes also result in cortical visual impairment.
A number of adverse perinatal events correlate with the development of PVL. Most important, PVL tends to occur
in the larger premature infant, with the highest incidence by both neuropathologic and ultrasound criteria being between 27 and 30 weeks’ gestational age (109,110) (Table 6.1). In the more recent French series of Baud and coworkers, published in 1999, the highest incidence (12.9%) was, however, seen in infants whose gestational age was 24 to 26 weeks (111). Other notable risk factors include prenatal factors such as premature, prolonged, or both premature and prolonged rupture of membranes, chorioamnionitis, and intrauterine infections (109,110,112). Several perinatal and postnatal factors also appear to be of importance. These are listed in Table 6.2 (4,113,114). In many instances, however, infants in whom PVL evolved had a relatively benign postnatal course (110). Although systemic hypotension has been suggested as an important pathogenetic factor, several studies failed to show an association between hypotension and PVL (115,116). In part, this lack of documentation reflects the lack of direct and continuous blood pressure recordings, or it might indicate that PVL results from a discrepancy between the metabolic requirements of periventricular white matter and its perfusion (117). We also stress that in infants of less than 31 weeks’ gestation, relatively small reductions in systemic blood pressure (less than 30 mm Hg) for 1 hour or longer suffice to induce cerebral infarcts (118). This is particularly true in those in whom autoregulation is defective or has been disrupted by asphyxia. As a rule, the less mature the periventricular vasculature, the less significant are the clinical complications that accompany the evolution of PVL.
in the larger premature infant, with the highest incidence by both neuropathologic and ultrasound criteria being between 27 and 30 weeks’ gestational age (109,110) (Table 6.1). In the more recent French series of Baud and coworkers, published in 1999, the highest incidence (12.9%) was, however, seen in infants whose gestational age was 24 to 26 weeks (111). Other notable risk factors include prenatal factors such as premature, prolonged, or both premature and prolonged rupture of membranes, chorioamnionitis, and intrauterine infections (109,110,112). Several perinatal and postnatal factors also appear to be of importance. These are listed in Table 6.2 (4,113,114). In many instances, however, infants in whom PVL evolved had a relatively benign postnatal course (110). Although systemic hypotension has been suggested as an important pathogenetic factor, several studies failed to show an association between hypotension and PVL (115,116). In part, this lack of documentation reflects the lack of direct and continuous blood pressure recordings, or it might indicate that PVL results from a discrepancy between the metabolic requirements of periventricular white matter and its perfusion (117). We also stress that in infants of less than 31 weeks’ gestation, relatively small reductions in systemic blood pressure (less than 30 mm Hg) for 1 hour or longer suffice to induce cerebral infarcts (118). This is particularly true in those in whom autoregulation is defective or has been disrupted by asphyxia. As a rule, the less mature the periventricular vasculature, the less significant are the clinical complications that accompany the evolution of PVL.
TABLE 6.1 Incidence of Periventricular Leukomalacia According to Gestational Age | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
TABLE 6.2 Factors Predisposing to the Evolution of White Matter Damage in Premature Infants | |
|---|---|
|
Several observational studies have reported that both maternal preeclampsia and the prenatal administration of magnesium resulted in a lower incidence of spastic diplegia, and by inference of PVL, in very low birth weight infants (119,120,121). These observations could not be confirmed in a controlled, retrospective study (122), and randomized clinical trials will be necessary to determine the effectiveness of magnesium. In addition, there are large and still unexplained differences among centers in the outcomes of extremely low birth weight infants, which also will require controlled clinical trials (123).
Newer imaging techniques such as ultrasonography and magnetic resonance imaging (MRI) permit the following of the evolution of PVL. In the series of infants autopsied by Iida and colleagues (90), the prenatal onset of PVL was observed in 20% of stillborn infants and in 16.4% of infants who died by 3 days of age. These findings have been confirmed by ultrasound studies showing the presence of cystic PVL as early as the third day of life (90,124). The evolution of PVL can be followed by ultrasonography. During the first week of life, transient hyperechoic periventricular areas are frequent and probably represent a persistent germinal matrix (125). Persistent echogenic foci are pathologic, however. They too are seen during the first week of postnatal life. Within 1 to 3 weeks they are replaced with echolucent, cystic foci (cystic leukomalacia) (see Fig. 6.7C and D). As the intracystic fluid becomes resorbed, these cysts disappear and are replaced by gliosis (126). PVL can be accompanied by cystic lesions in the subcortical white matter and by delayed myelination (127). In some instances, gliosis becomes interspersed with areas of microcalcification (128). Calcification is more likely when lesions are not extensive. Premature infants with PVL have a marked reduction in cerebral cortical gray matter volume at term as compared to premature infants without PVL. This may reflect destruction of corticopedal, corticofugal, and associative fibers with secondary impairment of neuronal differentiation (129).
Periventricular echodensities can reflect several neuropathologic entities aside from PVL. They also are observed in hemorrhagic infarctions such as are seen in association with IVH and in ischemic edema (130). PVL becomes hemorrhagic in up to 25% of infants (131), mostly a consequence of a hemorrhage into the ischemic area, the outcome of subsequent reperfusion (4).
Parasagittal Cerebral Injury
The most common site of brain damage in the term newborn is the cortex. Experimental studies have confirmed that the parasagittal cortex is the earliest and most severely damaged on prolonged asphyxia, with the amount of damage increasing geometrically with increasing duration of asphyxia (132). The lesions characteristically involve the territory supplied by the most peripheral branches of the three large cerebral arteries (Fig. 6.8) (133). Infarctions in this area are secondary to arterial or venous stasis and thromboses. One common pattern for the distribution of lesions, termed arterial “border zone” or “watershed lesions,” usually results from a sudden decrease in systolic blood pressure and cerebral perfusion. Watershed is a term first used in the early nineteenth century in England to describe the strip of land between two more or less parallel rivers or streams. This strip was protected from damming of one stream because it had an alternative water supply from the other, but it also was the most vulnerable and the first land to become parched during periods of general drought because it received the last water from both streams. Extrapolated to the cerebrovascular circulation, a watershed thus is the territory between two major arterial supplies, protected from occlusion of one of the arteries but vulnerable to an even transient period of systemic hypotension or hypoperfusion. The best-known watershed zones in the brain are between the anterior and middle cerebral artery circulations and between the middle and posterior cerebral circulations. Watershed zone infarcts are not infrequent in the neonate in these same regions that are affected in adults (Fig. 6.9), and usually denote a previous episode of systemic hypotension associated with fetal distress during late gestation or just prior to birth. Watershed zone infarcts in the full-term neonatal cerebrum are usually ischemic infarcts, but in about 30% of cases they are hemorrhagic. In preterm infants, hemorrhagic watershed infarcts are more frequent than ischemic infarcts.
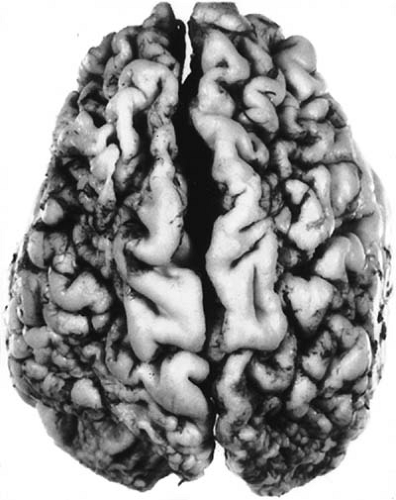 FIGURE 6.8. Watershed pattern in a 10-year-old child with history of prolonged labor and spastic quadriparesis. Symmetric atrophy is seen in border zones of anterior, middle, and posterior cerebral arteries. (From Lindenberg R. Compression of brain arteries as pathogenetic factor for tissue necrosis and their areas of predilection. J Neuropathol Exp Neurol 1955;14:223. With permission.) |
Another watershed zone occurs in the tegmentum of the brainstem. A series of 25 to 30 paired triads of vessels arise from the basilar artery; this series extends from the upper midbrain to the lower medulla oblongata. The three vessels of the triad on each side of the brainstem are (a) the paramedian penetrating artery, which extends dorsally next to the midline from the basilar artery to the floor of the fourth ventricle; (b) the short branches of the circumferential artery, which travels from the basilar artery around the outside of the brainstem to penetrate and supply the ventrolateral brainstem; and (c) the long branches of the circumferential artery, which encircles the brainstem and then penetrates dorsolaterally. The tegmentum of the pons and medulla is a watershed zone between the territories of the paramedian penetrating and the long circumferential arteries (Fig. 6.10) (133a).
Damage is maximal in the posterior parietal-occipital region, becoming less marked in the more anterior portions of the cortex. The lesions in the affected area can be located in the cortex or in the white matter. When gray matter is affected, damage usually involves the portions around the depth of the sulci. In part, this distribution can reflect the effect of cerebral edema on the drainage of the cortical veins, and, in part, it can be the consequence of the impoverished vascular supply of this area in the healthy human newborn. Bilateral parasagittal infarction also may result from sagittal sinus thrombosis, an event usually associated with infections (sepsis and meningitis) or with dehydration in young infants.
Lesions involving damage to the deeper portions of gray matter have been termed ulegyria (mantle sclerosis, lobar sclerosis, nodular cortical sclerosis) (3). A common abnormality, ulegyria accounts for approximately one-third of clinical defects caused by circulatory disorders during the neonatal period (133). Its characteristic feature is the localized destruction of the lower parts of the wall of the convolution, with relative sparing of the crown. This produces
a “mushroom” gyrus (Fig. 6.11). The margins of affected gray matter often contain abnormally dense aggregates of myelinated fibers, whereas adjacent white matter shows a considerable amount of myelin loss and compensatory gliosis (134). Later, coarse bundles of abnormally oriented, heavily myelinated fibers traverse the gliotic tissue, and the myelin sheaths enclose astrocytic processes as well as axons, similar to status marmoratus of the basal ganglia (135). Laminar necrosis of layer 3 often accompanies ulegyria.
a “mushroom” gyrus (Fig. 6.11). The margins of affected gray matter often contain abnormally dense aggregates of myelinated fibers, whereas adjacent white matter shows a considerable amount of myelin loss and compensatory gliosis (134). Later, coarse bundles of abnormally oriented, heavily myelinated fibers traverse the gliotic tissue, and the myelin sheaths enclose astrocytic processes as well as axons, similar to status marmoratus of the basal ganglia (135). Laminar necrosis of layer 3 often accompanies ulegyria.
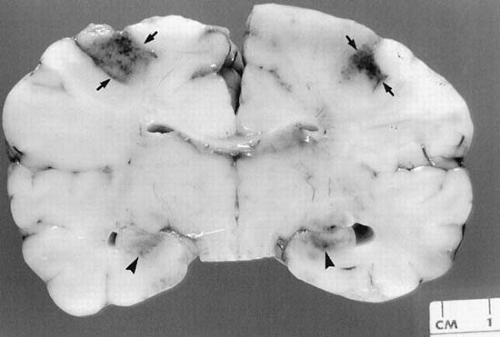 FIGURE 6.9. Coronal section of cerebrum, at the level of the thalamus and third ventricle, of a 35-week preterm infant who suffered multiple episodes of severe bradycardia during prolonged labor and lived only a few hours after birth. Bilateral watershed hemorrhagic infarcts are seen at the junctions between anterior cerebral and middle cerebral artery territories (arrows) and between the middle cerebral and posterior cerebral artery territories (arrowheads). The ventricles are normal and no periventricular hemorrhages are seen. |
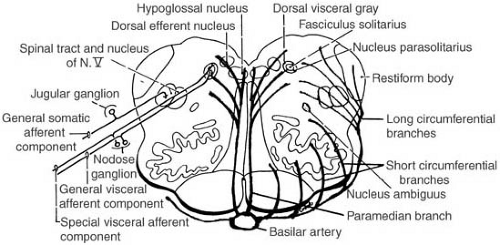 FIGURE 6.10. Drawing of a transverse section of the medulla oblongata to demonstrate the tegmental watershed zone, where the end-perfusions overlap from the parasagittal penetrating artery and the long circumferential artery, both arising from the basilar artery. Within this tegmental zone are the nucleus/tractus solitarius (respiratory center) and nucleus ambiguus (deglutition center) as well as several cranial nerve nuclei. (From Sarnat HB. Watershed infarcts in the fetal and neonatal brainstem. An aetiology of central hypoventilation, dysphagia, Möbius syndrome and micrognathia. Eur J Paediatr Neurol 2004;8:71–87. With permission.) |
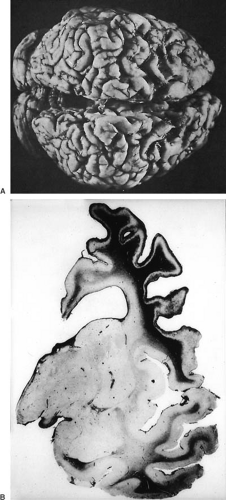 FIGURE 6.11. A: Lobar sclerosis (ulegyria) in a 5-year-old child with mental retardation and spastic since infancy. Sclerosis and distortion of the frontoparietal convolutions of the cerebrum are present. B: Coronal section of same brain showing shrunken and gliotic convolutions. The sulci are deepened and widened (Holzer’s stain for myelin fibers). (From Towbin A. The pathology of cerebral palsy. Springfield, IL: Charles C Thomas, 1960. With permission.) |
Ulegyria can be extensive or so restricted that the gross appearance of the brain is normal. It most commonly occurs in major arterial watershed border zones, in a parasagittal distribution in the venous drainage territory of the superior sagittal sinus, or within the territory of a major cerebral artery with partial occlusion (136). The perisulcal topography of the necrosis is related to reduced perfusion as a consequence of impaired sulcal venous drainage resulting from the compression of the stems of veins that ascend between gyri that have become edematous from previous hypoxic episodes (137). When ulegyria is widespread, an associated cystic defect in the subcortical white matter (porencephalic cyst) and dilatation of the lateral ventricles often occur. The meninges overlying the affected area are thickened and the small arteries occasionally can show calcifications in the elastica. Less often, ulegyria involves the cerebellum.
Marin-Padilla studied the postinjury gray matter alterations in ulegyria and found that the surviving cortex acquires a cortical dysplasia that affects the structural and functional differentiation of neurons, glial elements, and synaptic organization. Marin-Padilla proposed that the consequences of the acquired cortical dysplasia represent the main pathogenetic mechanism for epilepsy and other neurologic sequelae to perinatal brain damage (138).
Abnormalities of Basal Ganglia
Abnormalities within the basal ganglia are seen in the majority of patients subjected to perinatal asphyxia [84% in the series of Christensen and Melchior (139)]. One common lesion seen in this area has been termed status marmoratus. This picture was described first by Anton (140) in 1893 and later by the Vogt and Vogt (141). Fundamentally, the pathologic picture is one of glial scarring corresponding to the areas of tissue destruction. It is characterized by a gross shrinkage of the striatum, particularly the globus pallidus, associated with defects in myelination. Although in some cases myelinated nerve fibers, probably of astrocytic origin (135), are found in coarse networks resembling the veining of marble (hence the name of the condition, status marmoratus) (Fig. 6.12), in other cases the principal pattern is one of a symmetric demyelination (status dysmyelinatus) (142). Hypermyelination and demyelination probably represent different responses to the same insult. Hypermyelination probably results from oligodendrocytes becoming activated to produce excessive myelin, and, lacking enough axons to ensheath, they envelop astrocytic processes. The number of nerve cells in the affected areas is usually conspicuously reduced, with the smaller neurons in the putamen and caudate nucleus appearing more vulnerable. Cystic changes within the basal ganglia were stressed by Denny-Brown but are rarely extensive (143). Although the abnormalities within the basal ganglia are often the most striking, a variety of associated cortical lesions can be detected in most instances.
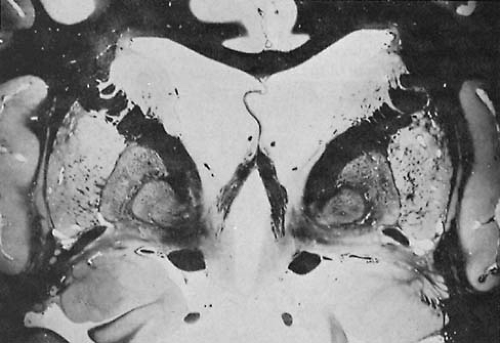 FIGURE 6.12. Status marmoratus of basal ganglia. (From Merritt HH. A textbook of neurology, 6th ed. Philadelphia: Lea & Febiger, 1979. With permission.) |
It has been our experience, as well as that of others, that this condition is the result of an acute, severe hypoxic insult (144). As a rule, the asphyxia is not as prolonged as that which results in multicystic encephalomalacia, a condition in which there is also extensive damage to the cerebral cortex and white matter. The reason for the selective vulnerability of the basal ganglia to asphyxia is alluded to in another section of this chapter.
Given that these regions have a higher baseline oxygen consumption than other regions of the brain, as has been evidenced by positron emission tomography (PET) (145), other factors also must be operative to account for the particular vulnerability of the putamen and anterior thalamus (146). The most recent thinking is that the topography of neuronal death points to a primary role of glutamate excitotoxicity in basal ganglia neuronal damage, with the type of cell death induced by asphyxia (necrosis or apoptosis) being determined by neuronal maturity and the severity and duration of asphyxia (147). Also playing a role is the density of excitatory receptors, the differential expression of ionotropic and metabotropic glutamate receptors, their differential sensitivities, and the expression of the various receptor subtypes (76,147). Variations in the subunit composition of the receptor and changes in the expression of glutamate receptors with maturation may also influence the sensitivity of neurons to perinatal asphyxia (147a). Because astrocytes and oligodendroglia also express glutamate receptors, these cells may participate in basal ganglia injury (147). The striatal gamma-aminobutyric acid (GABA)-ergic, medium-sized inhibitory neurons also are sensitive to asphyxia, whereas the striatal cholinergic interneurons are resistant to asphyxia (76). Basal ganglionic neurons expressing nitric oxide synthase participate in oxidative stress and excitotoxicity, which often lead to the death of neighboring cells (147b,148).
In infants who suffer basal ganglia necrosis secondary to asphyxia, neuronal immunoreactivity to the various glutamate receptors was consistently decreased, with the areas of decreased immune reactivity corresponding to the damaged regions (149). Neonatal asphyxia triggers a cascade of gene expressins for tyrosine hydroxylase and D1 and D2 dopamine receptors in experimental animals, but the importance of this finding is unclear (150).
Abnormalities of Cerebellum, Brainstem, and Pons
Occasionally, the major structural alterations resulting from perinatal injury are localized to the cerebellum. In the majority of instances, the involvement is diffuse, with widespread disappearance of the cellular elements of the cerebellar cortex, notably the Purkinje cells, and the dentate nucleus (3,151). As with the periventricular germinal matrix around the lateral ventricles, the external granular layer of the cerebellum is vulnerable to spontaneous hemorrhage, especially in preterm infants of young gestational age. However, in the vast majority of infants, selective cerebellar involvement is not the consequence of asphyxia (152).
In general, the human neonatal brainstem appears to be more resistant to ischemic and hypoxic insults than the cerebral cortex, but it is not invulnerable, and sometimes lesions are more prominent in the brainstem than in supratentorial structures. The lesions are usually symmetric and involve both gray matter nuclei and adjacent white matter tracts, but the gray matter is more focally involved, perhaps because of its higher metabolic rate. Lesions may involve the inferior or superior colliculi almost selectively (3), or infarction may occur in the central core of the brainstem or selectively in the periaqueductal gray matter (3,136,153,154). Such multiple deep microinfarcts at times extend rostrally to involve deep supratentorial structures such as the thalamus and corpus striatum (155). Bilateral tegmental infarcts of the pons and medulla oblongata in
particular are frequent sequelae of transient fetal hypotension because the tegmentum is a watershed zone, futher discussed later (133a). Because of the microscopic size of many of the deep infarcts, particularly those of the brainstem, they are difficult to identify on neuroimaging. Nevertheless, some older lesions that occurred in fetal life several weeks before delivery may be visualized in CT by their calcification in the floor of the fourth ventricle (133a,155a). MRI occasionally demonstrates infarcts in the tegmentum or floor of the fourth ventricle if they are large enough (155b).
particular are frequent sequelae of transient fetal hypotension because the tegmentum is a watershed zone, futher discussed later (133a). Because of the microscopic size of many of the deep infarcts, particularly those of the brainstem, they are difficult to identify on neuroimaging. Nevertheless, some older lesions that occurred in fetal life several weeks before delivery may be visualized in CT by their calcification in the floor of the fourth ventricle (133a,155a). MRI occasionally demonstrates infarcts in the tegmentum or floor of the fourth ventricle if they are large enough (155b).
Infarction in the nuclei of the brainstem and thalami, induced in experimental animals by total asphyxia of 10 to 25 minutes’ duration, also can be seen in asphyxiated human infants with a history of acute profound asphyxial damage (156). Additionally, transient compression of the vertebral arteries in the course of rotation or hyperextension of the infant’s head during delivery can be a cause for circulatory lesions of the brainstem (157,158).
Involvement of the nucleus ambiguous, just ventrolateral to the tractus solitarius, can result in dysphagia because this nucleus provides motor neurons for the muscles of deglutition. Involvement of the trigeminal motor nucleus by tegmental infarcts may damage motor neurons to the masticatory muscles, such as the masseter and pterygoids, and, in late gestation, the lack of function of these muscles leads to a lack of stimulus to growth of the mandible, so that micrognathia and even ankylosis of the temporomandibular joint is present by the time of birth (133a). Because the paired triads of vessels from the basial artery are 25 to 30 in number, from the midbrain to the caudal end of the medulla oblongata, the watershed tegmental infarcts are columnar and may extend posteriorly to even involve the hypoglossal nucleus so that the infant has atrophy and fasciculations of the tongue that may be mistaken for spinal muscular atrophy (133a,155c).
Neurosensory hearing loss after basilar artery insufficiency has both central and peripheral bases: The lateral lemniscus is at the margin of the tegmental watershed zone and the inferior colliculus is one of the most constant regions infarcted; the stria vascularis and hair cells of the cochlea receive their arterial supply from the basilar artery via the inferior anterior cerebellar artery (IACA). The labyrinth (semicircular canals) also receives arterial blood from the IACA, but its cells seem more resistant to irreversible injury from ischemia.
Table 6.3 lists the lesions and clinical expression seen with tegmental infarcts, but only rarely does an infant exhibit all deficits. Though much more frequent in fetuses and neonates than at any other time in life, tegmental watershed infarcts also can occur in older children and adults.
Unilateral tegmental infarcts also can involve the fetal and neonatal brainstem but are rare; such asymmetric lesions are more likely the result of a vascular anomaly of the basilar artery or birth trauma to the veretebral arteries than of generalized hypoperfusion of the brainstem due to transient shock in the fetus or neonate (133a,158).
TABLE 6.3 Clinical Sequelae of Tegmental Watershed Infarcts | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Pontosubicular Degeneration
Pontosubicular degeneration in isolation or accompanied by widespread cerebral damage has been described in premature and term infants (3,156,157). It is not a rare entity. It has been demonstrated in up to 59% of infants born before 38 weeks’ gestation who died in the first month of postnatal life, which suggests that it is the most common cerebral lesion of preterm neonates, exceeding even germinal matrix hemorrhages (4,159,160,161).
The condition represents a unique topology of pathologic neuronal apoptosis in the fetal and neonatal brain following hypoxia or ischemia. As its descriptive name implies, it selectively involves relay nuclei of the corticopontocerebellar pathway in the basis pontis and the subiculum, a transitional cortex between the three-layered hippocampus and the six-layered hippocampal gyrus. Although it may coexist with other hypoxic lesions in the cortex, thalamus, and cerebellum, these other regions are disproportionately less severely involved than the pontine nuclei and subiculum (136,159,162). Usually, there are no lesions in the tegmentum of the pons, although in rare instances symmetric microinfarcts have been described (163,164). Focal white matter infarcts also occur occasionally.
This distribution of infarcts is generally seen in infants older than 29 weeks’ gestation, most commonly in infants of 32 to 36 weeks’ gestation. The pattern can also occur in stillborns (165) and term infants and has occasionally been encountered in adult brains (166). The reason that pontosubicular degeneration is not better recognized by clinicians is that it is difficult to demonstrate during life and remains essentially a postmortem neuropathologic diagnosis.
The combination of infarcts in the basis pontis and the subiculum is difficult to explain because these regions do not share common afferent or efferent fiber connections, use different neurotransmitters, have morphologically
different types of neurons, arise from different embryologic primordia, and belong to different functional systems of the brain.
different types of neurons, arise from different embryologic primordia, and belong to different functional systems of the brain.
The pathogenesis of pontosubicular degeneration is poorly understood. Hyperoxemia in the presence of acidosis and hypoxia is described in some infants, suggesting that oxygen toxicity plays a role (160), but these cases represent only a small minority. Pontine neurons exhibiting karyorrhexis are immunoreactive to ferritin if accompanied by spongy changes and gliosis, suggesting that iron may be released to the damaged pontine neurons (167). In situ DNA fragmentation studies indicate apoptosis rather than frank necrosis as the mechanism of cellular death (168,169).
Pontosubicular degeneration has been described in stillborn fetuses, indicating that the lesions may result from intrauterine fetal distress and are not necessarily acquired intrapartum or postpartum (165,170,171). Sarnat provided details of the histologic progression of changes in the degenerating neurons (136).
Spinal Cord Lesions
The spinal cord is also vulnerable to hypoxic-ischemic injuries, and damage to anterior horn cells results from hypoperfusion of the watershed area between the vascular distribution of the anterior spinal and the dorsal spinal arteries (172). The resultant hypotonia is generally attributed to cerebral injury, but electromyography (EMG) can demonstrate a lower motor neuron injury.
Infarcts
An infarct, the consequence of a focal or generalized disorder of cerebral circulation that occurs during the antenatal or early postnatal period and acts in isolation, is a relatively rare cause of brain damage. In the series of autopsies studied by Barmada and colleagues, arterial infarcts were seen in 5.4%, and infarcts of venous origin were found in 2.4% (173). Almost 90% are unilateral, and in 83% the infarct is in the distribution of the middle cerebral artery (4). It is presumed to be the result of embolization arising from placental infarcts or of thrombosis caused by vascular maldevelopment, sepsis, or, as in the case of a twin to a macerated fetus, the exchange of thromboplastic material from the dead infant (3). In the series of Fujimoto and colleagues, 22% followed perinatal asphyxia; their onset was in the first 3 days of life (174). Infarcts can be asymptomatic or present with convulsions. Of 90 term infants presenting with seizures solely within 72 hours of birth, Cowan et al. found that 35 (39%) had focal cerebral infarction in arterial or parasagital distribution (30). Such infarctions are generally thought to be unrelated to intrapartum difficulties or the presence of neonatal encephalopathy, although the incidence of antenatal and intrapartum problems is higher in this group of infants than in a control population (175). Sreenan and coworkers had a different experience (176). In their series neonatal encephalopathy was encountered in 56% of infants who developed cerebral infarctions, and various adverse perinatal events occurred in about 75%. The presence of an inherited thrombophilic abnormality increased the risk of focal infarction. In the same population Mercuri and coworkers found that 30% of infants with focal infarction had a thrombophilic abnormality, usually heterozygosity for factor V Leiden or a high factor VIII concentration (177). In the series of Golumb and coworkers (178), 70% of infants with ischemic stroke who did not suffer from neonatal encephalopathy had anticardiolipin antibodies. Golumb reviewed the various prothrombotic disorders that contribute to the development of neonatal thrombotic infarcts (179).
Focal infarctions are less common in the premature than the term infant, and compared with the term infant the premature infant has a better prognosis with regard to neurologic residua (180).
Porencephaly
A porencephalic cyst is a large intraparenchymal cyst that always communicates with the ventricular system. Loculated cysts entirely within the subcortical white matter are not porencephalic and are pseudocysts rather than true cysts because they lack an epithelial lining. Porencephalic cysts are partly, though usually sparsely, covered by ependyma. Porencephaly results from infarction in the territory of a major artery, usually the middle cerebral artery, although at times it may be a sequel to a grade 4 intraventricular hemorrhage (IVH) that extends the ventricle lumen into the empty parenchymal space left by the reabsorption of the hematoma. It is not a watershed infarct.
Porencephalic cysts often appear to communicate also with the overlying subarachnoid space, and such a communication may be reported by radiologists, but careful neuropathologic examination demonstrates that a thin pial membrane and sometimes arachnoidal tissue separate the porencephalic and subarachnoid compartments. (Fig. 6.13). The pia derives its vascular supply from meningeal rather than cerebral vessels. This membrane is too thin to resolve by CT or MRI, but is important during life in terms of fluid shifts and CSF flow.
The cerebral cortex immediately surrounding a porencephalic cyst often appears to be polymicrogyric. This is secondary to ischemia and atrophy of immature gyri and should not be misconstrued as a primary dysgenesis. These small gyri are gliotic and have extensive neuronal loss.
Porencephaly is usually limited to one hemisphere, and the clinical correlates are spastic hemiplegia, hemisensory deficits, and often hemianopia. Porencephalic cysts following grade 4 IVH are visualized in survivors 10 days to 8 weeks after the event (184). Although it is not a
progressive lesion and does not obstruct the flow of CSF in the chronic phase, the porencephalic cyst occasionally enlarges and causes symptoms of intracranial hypertension. The reason for this phenomenon is the pulsation of the choroid plexus, which may induce a “waterhammer effect” because the force of the pulsations is transmitted to a larger surface area and the resistance to stretch is therefore less. The choroid plexus does not derive its blood supply from the middle cerebral artery; hence it usually survives the infarct. In some cases, a ventriculoperitoneal shunt may be required to prevent further enlargement of the porencephalic cyst, encroachment on functional brain, and midline shift.
progressive lesion and does not obstruct the flow of CSF in the chronic phase, the porencephalic cyst occasionally enlarges and causes symptoms of intracranial hypertension. The reason for this phenomenon is the pulsation of the choroid plexus, which may induce a “waterhammer effect” because the force of the pulsations is transmitted to a larger surface area and the resistance to stretch is therefore less. The choroid plexus does not derive its blood supply from the middle cerebral artery; hence it usually survives the infarct. In some cases, a ventriculoperitoneal shunt may be required to prevent further enlargement of the porencephalic cyst, encroachment on functional brain, and midline shift.
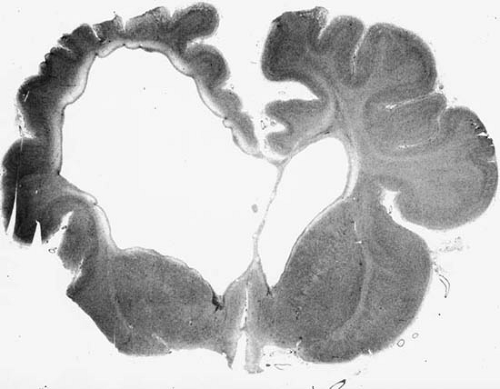 FIGURE 6.13. Porencephaly in middle cerebral artery distribution of a 4-month-old boy born at 30 weeks’ gestation and who suffered fetal distress for more than a week before delivery. This coronal section of the brain at autopsy shows a large cavity replacing most of the right cerebral hemisphere and continuous with the lateral ventricle on that side. A thin ribbon of white matter and a thin cortex remain, but there is polymicrogyria of the right hemisphere in response to the atrophy. The left, “good” hemisphere shows pachygyria because the convolutional development of the brain became arrested at the time of onset of the fetal distress. Hematoxylin-eosin. ×1.5 (original magnification). |
Rarely, porencephaly is transmitted as an autosomal dominant disorder with incomplete penetrance. The presentation is with variable degrees of hemiparesis, seizures, and mental retardation (185). A thrombotic event during late pregnancy is believed to be responsible. The gene has been mapped to chromosome 13qter (186).
Intracranial Hemorrhage
Whereas mechanical trauma can be responsible for a subdural hemorrhage and, less commonly, a primary subarachnoid hemorrhage, it plays a relatively unimportant role in the evolution of periventricular-intraventricular hemorrhage (IVH), the most common form of neonatal intracranial hemorrhage (Table 6.4) (4,187,188). The various grades of hemorrhage are defined in Table 6.5 and depicted in Figs. 6.14, 6.15, and 6.16.
The site of the bleeding that results in an IVH is determined by the maturity of the infant. In the premature
infant, bleeding originates in the capillaries of the subependymal germinal matrix, usually over the body of the caudate nucleus (188). With increasing maturation the germinal matrix involutes, so that in the term infant the choroid plexus becomes the principal site of the hemorrhage (114,188). Although Hayden and coworkers encountered IVH in 4.6% of term neonates (189), its incidence increases markedly with decreasing maturity, so that when ultrasonography is performed on infants with birth weights less than 1,500 g, a hemorrhage can be documented in as many as 50%. A high-grade hemorrhage is more common in this group than in infants with birth weights greater than 1,500 g (190,191,192). At times, even premature infants of advanced gestational age may have extensive hemorrhages that can destroy the thalamus or basal ganglia (Figs. 6.3 and 6.15).
infant, bleeding originates in the capillaries of the subependymal germinal matrix, usually over the body of the caudate nucleus (188). With increasing maturation the germinal matrix involutes, so that in the term infant the choroid plexus becomes the principal site of the hemorrhage (114,188). Although Hayden and coworkers encountered IVH in 4.6% of term neonates (189), its incidence increases markedly with decreasing maturity, so that when ultrasonography is performed on infants with birth weights less than 1,500 g, a hemorrhage can be documented in as many as 50%. A high-grade hemorrhage is more common in this group than in infants with birth weights greater than 1,500 g (190,191,192). At times, even premature infants of advanced gestational age may have extensive hemorrhages that can destroy the thalamus or basal ganglia (Figs. 6.3 and 6.15).
TABLE 6.4 Major Types of Neonatal Intracranial Hemorrhage and Usual Clinical Setting | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 6.5 Grading of Severity of Periventricular-Intraventricular Hemorrhage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
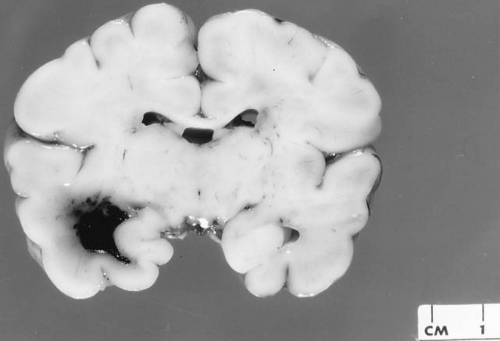 FIGURE 6.14. Intraventricular hemorrhage, grade 2. Focal hemorrhage of the germinal matrix breaking through the ependyma and causing focal intraventricular hemorrhage that does not extend throughout or dilate the ventricular system. The coronal section of this brain of a 32-week premature infant shows hemorrhage into the left temporal horn and local dilatation of that horn, but blood does not extend into the frontal horn or across the midline, and the rest of the ventricular system is not dilated. In the dorsolateral periventricular region of the involved temporal horn, hemorrhage is seen in the parenchyma, but confined to that zone. |
The pathogenesis of IVH is not completely understood. The predisposition of the premature infant to IVH is in part due to the presence of a highly vascularized subependymal germinal matrix, to which a major portion of the blood supply of the immature cerebrum is directed. Furthermore, the capillaries of the premature infant have less basement membrane than those of the mature brain, and there is a paucity of tight junctions that is compounded by incomplete coverage of blood vessels by astrocytic end feet leading to fragility of the blood vessels. Finally, abnormalities in the autoregulation of arterioles in premature and distressed term infants impair the infants’ response to hypoxia and hypercarbia and thus permit transmission of arterial pressure fluctuations to the fragile periventricular capillary bed.
Prenatal as well as perinatal and postnatal factors have been implicated in the evolution of IVH. Lack of adequate matching for gestational age and birth weight have, however, confounded many results (193). As a rule, IVH that develops in the first 12 hours of life is associated with variables relating to labor and delivery, whereas IVH that starts
later is associated with postpartum variables. Premature rupture of membranes and maternal chorioamnionitis increase the risk for the condition, suggesting that cytokines play a role in its evolution (192,194,195).
later is associated with postpartum variables. Premature rupture of membranes and maternal chorioamnionitis increase the risk for the condition, suggesting that cytokines play a role in its evolution (192,194,195).
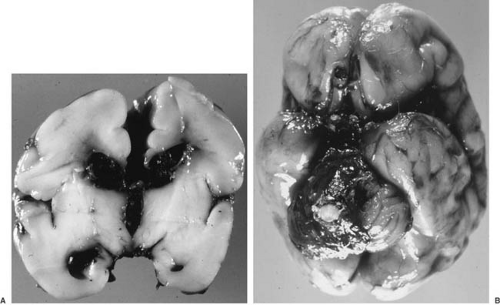 FIGURE 6.15. A: Intraventricular hemorrhage, grade 3. More extensive intraventricular hemorrhage than in Fig. 5.2, in a 28-week premature infant. Hemorrhage extends throughout the ventricular system and causes dilatation of the lateral ventricles bilaterally and the third ventricle; the original site of origin of the hemorrhage may be difficult to identify, but periventricular leukomalacia is seen around both frontal horns and the left side of the third ventricle. B: Blood from the lateral and third ventricles is seen in the subarachnoid space at the base of the brain, having exuded through the aqueduct and fourth ventricle and out the foramina of Luschka and Magendie during the course of cerebrospinal fluid flow. |
In most infants, acute fluctuations in cerebral blood flow and an impaired cerebral vascular autoregulation are more important than prenatal factors in the evolution of an IVH. Clinical studies have shown that infants with intact cerebrovascular autoregulation are at low risk for IVH. In contrast, a variety of adverse factors that disrupt autoregulation are associated with a high risk of IVH. These include low Apgar scores, respiratory distress, artificial ventilation, the presence of a patent ductus arteriosus, and the various complications of perinatal and postnatal asphyxia (192,194,196). An elevation of venous pressure also has been implicated. Such an elevation can occur in the course of labor and delivery, or it can accompany positive-pressure ventilation, pneumothorax, hypoxic-ischemic myocardial failure, or hyperosmolality induced by administration of excess sodium bicarbonate (4).
The importance of pneumothorax caused by positive-pressure ventilation in producing IVH was stressed by McCord and coworkers, who were able to reduce the incidence of respiratory distress syndrome and with it the incidence of IVH by treatment with surfactant (197). In addition to surfactant, maternal tocolysis using ritrodrine, early low-dose indomethacin, and antenatal steroid treatment also reduce the incidence of IVH in very low birth weight premature infants (193,198,199,200). Neonatal risk factors that predispose to the evolution of IVH include clotting disorders, reduced hemoglobin, hypercarbia, and hypoglycemia (4).
In the premature infant, the hemorrhage does not occur at the time of delivery but tends to commence later, most commonly 24 to 48 hours after a major asphyxial insult, whether at the time of birth or subsequently (192,201). In the experience of Ment and associates, 74% of hemorrhages were detected by ultrasonography within 30 hours after birth (202) (Fig. 6.16). In the series of Trounce and colleagues, 15% of infants developed an IVH after 2 weeks
of age (191). In some infants bleeding can be a slow process rather than a sudden event (203). The extent of the hemorrhage can range from a slight oozing to a massive intraventricular bleed with an associated asymmetric periventricular hemorrhagic infarction and an extension of the blood into the subarachnoid space of the posterior fossa (see Figs. 6.14 and 6.15) (204).
of age (191). In some infants bleeding can be a slow process rather than a sudden event (203). The extent of the hemorrhage can range from a slight oozing to a massive intraventricular bleed with an associated asymmetric periventricular hemorrhagic infarction and an extension of the blood into the subarachnoid space of the posterior fossa (see Figs. 6.14 and 6.15) (204).
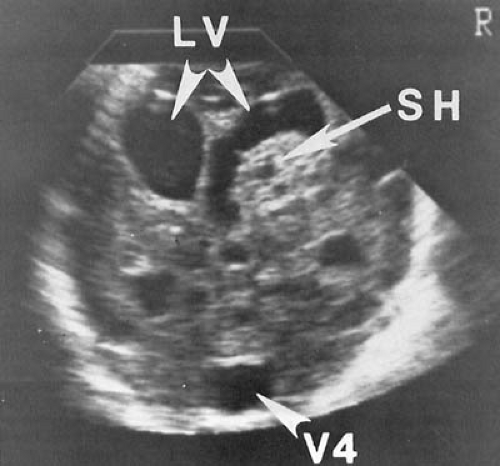 FIGURE 6.16. Intraventricular hemorrhage in a 1,400-g infant of 30 weeks’ gestation who suffered birth asphyxia. Coronal ultrasonographic scan reveals moderate hydrocephalus and a large subependymal hemorrhage (SH) in the wall of the right lateral ventricle (LV, lateral ventricles; V4, fourth ventricle). (From Babcock DS, Han BK. The accuracy of high-resolution, real-time ultrasonography of the head in infancy. Radiology 1981;139:665. With permission.) |
Blood usually clears rapidly from the intraventricular and subarachnoid spaces. In fact, hemosiderin deposition, a reliable and permanent neuropathologic marker of old hemorrhage in the adult brain, is found rarely in children’s brains after an IVH. Despite the resolution of the fresh blood, brain injury is a relatively common result of IVH. Several mechanisms are believed to play a role (4).
The injury can be the result of an antecedent asphyxial injury that predisposed the infant to the bleeding.
In the presence of a large IVH, intracranial pressure will increase, which, in turn, will reduce cerebral perfusion.
The IVH can induce arterial vasospasm. As demonstrated by PET, cerebral blood flow becomes abolished in the area of an intraparenchymal hematoma and is reduced twofold to threefold over the entire affected hemisphere (205). How a hemorrhage induces such widespread vasospasm is unclear. Like the vasospasm encountered in older children and adults after a subarachnoid hemorrhage induced by the rupture of an aneurysm (see Chapter 13), the vasospasm could be related to the presence of high concentrations of blood in the CSF (206). Vasospasm may well have its major effect on the middle cerebral artery; as judged from the pulsatility index in the anterior cerebral artery, Perlman and Volpe found no consistent effect of an IVH on flow in the anterior cerebral artery (207).
Metabolic alterations are responsible for subsequent neurologic abnormalities. Cerebral glucose metabolism is markedly reduced (208), and, as determined by MR spectroscopy, the brain phosphocreatine concentration is reduced for several weeks after the hemorrhage (209).
Unilateral or grossly asymmetric destruction of periventricular white matter can be the result of an ischemic reperfusion injury. A fan-shaped hemorrhagic infarct, visualized by ultrasonography as an intracerebral periventricular, echodense lesion, is not unusual and can be demonstrated in approximately 15% of infants with IVH and in approximately one-third of those who harbor a severe hemorrhage (4,210) (Fig. 6.17). It is marked by a large region of hemorrhagic necrosis in the periventricular white matter at the point where the medullary veins become confluent and join the terminal vein in the subependymal region. The necrosis is usually markedly asymmetric; it is unilateral in the majority of instances (96). Approximately 80% of cases are accompanied by a large IVH, and, in the past, the
infarction was mistakenly described as a parenchymal extension of the IVH. Periventricular hemorrhagic infarction is believed to result from the IVH compressing and obstructing the terminal veins and interfering with their drainage (211). The periventricular hemorrhagic infarct produces tissue destruction and formation of cystic cavities and is associated with a poor functional outcome. Porencephalic cysts can develop in survivors and are visualized in 10 days to 8 weeks after the event (212).
FIGURE 6.17. Intracerebral hemorrhage in a neonate. Coronal ultrasonographic scan. The arrow indicates the presence of the hematoma. Displacement of the ventricular system occurred. (Courtesy of Dr. Eric E. Sauerbrei, Kingston General Hospital, Kingston, Ontario, Canada.)
White matter injury can also result from the toxic effects of proinflammatory cytokines or the release of a variety of reactive oxygen species (213). Iron released during heme catabolism has the potential of generating hydroxyl radicals and inducing oxidative damage on immature oligodendroglia.
Progressive ventricular dilatation is a common sequel to IVH (Fig. 6.18). Evolving 1 to 3 weeks after the hemorrhage, it is caused by a fibrotic reaction that obliterates the subarachnoid spaces and induces ventricular dilatation with or without increased intracranial pressure (normal-pressure hydrocephalus) (4). The factors responsible for normal-pressure hydrocephalus in the neonate are poorly understood.
When an IVH occurs in term infants, it generally emanates from the choroid plexus, less frequently from the subependymal germinal matrix. The major causes are trauma and perinatal asphyxia (4). In the experience of Volpe, approximately one-half of term newborns with IVH experienced difficult deliveries. The experience of Palmer and Donn is similar (214). In approximately 25% of cases, the IVH is of unknown etiology. One cause that probably accounts for a large number of these cases is a cryptic hemangioma of the choroid plexus, well demonstrated at autopsy (215,216). The lesions range in size from thin-walled cavernous angiomas to fully formed arteriovenous malformations. They are sometimes difficult to show by imaging, in part because the hemorrhage may destroy the original vascular malformation or obscure its remains (216). Unruptured angiomas of the choroid villi are not uncommon as incidental findings at autopsy and must be distinguished from simple vascular congestion. It is a relatively common complication of Sturge-Weber syndrome (see Chapter 12). One of us (H.S.) has found them to be more frequent in patients with cerebral malformations or chromosomal abnormalities.
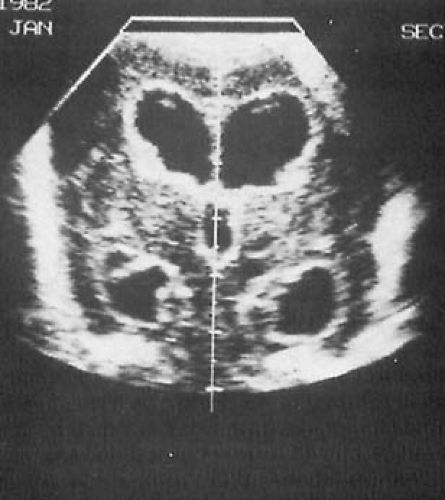 FIGURE 6.18. Posthemorrhagic hydrocephalus in a 2-week-old premature infant. The lateral ventricles and the third ventricle are dilated; the fourth ventricle is not visualized. The increased echogenicity of the ventricular wall indicates posthemorrhagic hydrocephalus, as distinct from hydrocephalus resulting from a malformation, in which the hyperecho ring is absent. (Courtesy of Dr. W. Donald Shields, Division of Pediatric Neurology, University of California, Los Angeles, CA.) |
Term infants with IVH tend to become symptomatic at a later age, often not until the fourth week of life. Irritability, changes in alertness, and seizures are common presenting symptoms. In the series of Palmer and Donn, seizures were the first symptom in 69% of cases; 23% of infants presented with apnea (214).
Extension of intracranial hemorrhage to the spinal canal is more frequent than is recognized. Both epidural and subdural bleeding have been encountered, the former being far more common. Epidural bleeding is associated not only with intracranial hemorrhage owing to asphyxia, but also with traumatic birth injuries (217,218). Although these hemorrhages either are asymptomatic or induce deficits that are obscured by the more obvious symptoms of an intracranial hemorrhage, diagnosis by MRI of the spinal cord is now possible.
Primary and Secondary Malformations of the Central Nervous System
In addition to direct trauma, asphyxia, and circulatory disturbances, malformations of the CNS play an important part in the genesis of the lesions of perinatal asphyxia and trauma. Little doubt exists that in the premature infant, for instance, both faulty maturation of the nervous system and a greater vulnerability to perinatal trauma and asphyxia are responsible for the high incidence of neurologic deficits (Table 6.6) (219). The relative frequency of prenatal and perinatal brain lesions in individuals with moderate or severe mental retardation can be determined from autopsy studies such as those by Freytag and Lindenberg (133) (Table 6.7) or from MRI evaluation of children with mental retardation and/or cerebral palsy (31).
Ischemic, hypoxic, and traumatic insults of the fetal brain in the second and third trimesters can induce malformations that are not primary defects of genetic
programming. Because development is incomplete, lesions that interrupt or alter radial glial fibers, for example, can prevent further neuroblast and glioblast migration before the process is complete and may result in focal dysplasias of cortical lamination and in deep heterotopia of neurons arrested in migration (Fig. 6.19). The abnormal synaptic relations that result from the abnormal anatomic positions of neurons may become a basis for later epilepsy (138).
programming. Because development is incomplete, lesions that interrupt or alter radial glial fibers, for example, can prevent further neuroblast and glioblast migration before the process is complete and may result in focal dysplasias of cortical lamination and in deep heterotopia of neurons arrested in migration (Fig. 6.19). The abnormal synaptic relations that result from the abnormal anatomic positions of neurons may become a basis for later epilepsy (138).
TABLE 6.6 Neuropathology in Premature and Full-Term Infants with Cerebral Palsy | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 6.7 Frequency of Brain Lesions | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Clinical Manifestations of Cerebral Perinatal Injuries
This section describes the clinical appearance of the neonate who has been subjected to perinatal asphyxia or trauma. It also traces the evolution of the spastic and extrapyramidal deficits and concludes with a discussion of the various syndromes of cerebral palsy, acknowledging that in many instances cerebral malformations play an etiologic role equaling or surpassing that of perinatal asphyxia and trauma.
The interested reader is referred to the pioneering studies by Paine on the evolution of tone and postural reflexes in neurologically damaged neonates (220,221).
Neonatal Period
The degree of a newborn’s functional abnormality secondary to asphyxia incurred during labor and delivery depends on the severity, timing, and duration of the insult. (See the Introduction chapter for a description of the essentials of a neurologic examination of the infant or small child.)
After birth, the infant subjected to perinatal asphyxia shows certain alterations in alertness, muscle tone, and respiration. These important clinical features of HIE were first graded by Sarnat and Sarnat (222) (Table 6.8). Several
other grading schemes have been devised. In essence, they are similar to that of Sarnat and Sarnat; however, some schemes place infants with repetitive or prolonged seizures into grade 3 rather than grade 2 (22). Sarnat and Sarnat specifically excluded seizures as a criterion for grading acute encephalopathy because of poor correlation with outcome in their experience and because some of the most severely involved infants in stage 3 do not have seizures because the cerebral cortex is so severely impaired that it can no longer generate epileptic activity. They did include electroencephalographic (EEG) criteria, however, as a measure of cerebral function rather than of paroxysmal activity. They also emphasized the importance of autonomic features, with sympathomimetic effects in stage 1 and strong parasympathetic (i.e., vagal) effects in stage 2.
other grading schemes have been devised. In essence, they are similar to that of Sarnat and Sarnat; however, some schemes place infants with repetitive or prolonged seizures into grade 3 rather than grade 2 (22). Sarnat and Sarnat specifically excluded seizures as a criterion for grading acute encephalopathy because of poor correlation with outcome in their experience and because some of the most severely involved infants in stage 3 do not have seizures because the cerebral cortex is so severely impaired that it can no longer generate epileptic activity. They did include electroencephalographic (EEG) criteria, however, as a measure of cerebral function rather than of paroxysmal activity. They also emphasized the importance of autonomic features, with sympathomimetic effects in stage 1 and strong parasympathetic (i.e., vagal) effects in stage 2.
 FIGURE 6.19. Drawing of a coronal section of the cerebral hemisphere of a preterm infant to illustrate three possible sites where ischemic-hypoxic lesions might disrupt radial glial cells or their fibers to interfere with neuroblast and glioblast migration and thus cause secondary, acquired malformations as focal cortical dysplasias and subcortical heterotopia of incompletely migrated cells: (1) In the periventricular region, periventricular leukomalacia or grade 1 or 2 germinal matrix hemorrhage; (2) in the deep subcortical white matter, as either ischemic or hemorrhagic infarction; and (3) at the pial surface, where injury might cause retraction of radial glial end-feet. Examples of insults at the pial surface include contusions of the brain at delivery, subarachnoid hemorrhage, and neonatal meningitis. (From Sarnat HB. Cerebral dysgenesis. Embryology and clinical expression. New York: Oxford University Press, 1992. With permission.) |
TABLE 6.8 Clinical Features of Hypoxic-Ischemic Encephalopathy | ||
|---|---|---|
|
In the experience of Levene and coworkers, 3.9 in 1,000 newborn term infants develop grade 1 HIE, 1.1 in 1,000 develop grade 2 HIE, and 1.0 in 1,000 develop grade 3 HIE (223).
Infants with grade 1 HIE are irritable with some degree of feeding difficulty and are in a hyperalert state in which their eyes are open with a “worried” facial appearance and a decreased frequency of blinking. They seem hungry and respond excessively to stimulation. Tremulousness, especially when provoked by abrupt changes of limb position or tactile stimulation, can resemble seizures. Mild degrees of hypotonia can be documented by a head lag and a lack of the normal biceps flexor tone in the traction response from the supine position. The Landau reflex is often abnormal, in that the infant’s body tends to collapse into an inverted U shape.
A greater hypoxic insult results in the evolution of grade 2 HIE. Infants are lethargic or obtunded with delayed or incomplete responses to stimuli. Focal or multifocal seizures are common. Severely asphyxiated infants develop clinical signs of grade 3 HIE. The infant is markedly hypotonic. The sucking and swallowing reflexes are absent, producing difficulties in feeding. Palmar and plantar grasps are weak, the Moro reflex can be absent, and the placing and stepping reactions are impossible to elicit.
A variety of respiratory abnormalities can be encountered. These include a failure to initiate breathing after
birth, which suggests a hypoxic depression of the respiratory reflex within the brainstem. Tachypnea or dyspnea in the absence of pulmonary or cardiac disease also suggests a neurologic abnormality. Periodic bouts of apnea are normal in the smaller premature infant. In larger infants, periodic apnea can result from a depression of the respiratory reflex or can indicate a seizure disorder (see Chapter 14).
birth, which suggests a hypoxic depression of the respiratory reflex within the brainstem. Tachypnea or dyspnea in the absence of pulmonary or cardiac disease also suggests a neurologic abnormality. Periodic bouts of apnea are normal in the smaller premature infant. In larger infants, periodic apnea can result from a depression of the respiratory reflex or can indicate a seizure disorder (see Chapter 14).
The Apgar score has been used to measure the severity of the initial insult. Although in the past the score was used to determine the presence of a hypoxic insult, to do so is a misapplication. For instance, in the experience of Sykes and coworkers, only 20% of infants with a 5-minute Apgar score of less than 7 had an umbilical artery pH below 7.10 (224). Conversely, virtually all of the infants of 35 to 36 weeks’ gestation with a cord pH below 7.25 had a 5-minute Apgar score of 7 or more. In preterm infants the Apgar score is of even less value, and the more premature the infant, the more likely it is that the Apgar score will be low in the presence of a normal cord pH (225). Instead, the value of the Apgar score lies in being a simple and valuable predictor for survival during the neonatal period and for the ultimate outcome. In the series of Casey and coworkers, the neonatal mortality for term infants with a 5-minute Apgar score of 0 to 3 was 244 per 1,000, as contrasted with a mortality of 0.2 per 1,000 for intants with a 5-minute Apgar score of 7 to 10 (226). The extended Apgar scores (Apgar scores taken at 10 and 20 minutes of life), however, are even more valuable in predicting neurologic outcome, in that the likelihood of ensuing cerebral palsy increases significantly once the Apgar score remains under 3 for 10 minutes or longer (227). Moster and colleagues (228) noted that infants with 5-minute Apgar scores of 0 to 3 had an 81-fold increased risk for cerebral palsy compared with infants who had scores of 7 to 10. Term infants who had 5-minute Apgar scores of 0 to 3 and who also had signs consistent with neonatal encephalopathy had a significantly increased risk for impaired minor motor skills, reduced performances in reading and mathematics, and a variety of behavioral problems (229).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


