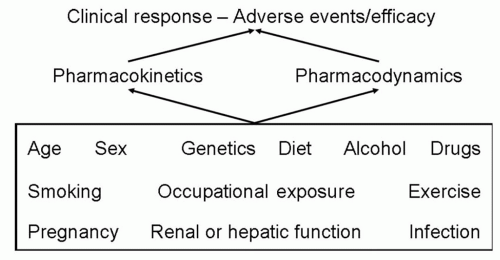
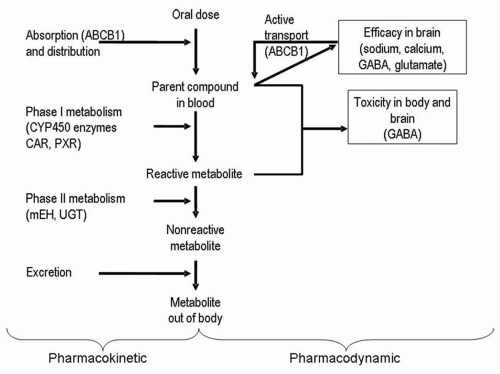
Metabolism of AEDs is divided into two phases (
10). Phase I reactions can be oxidative (mediated by cytochrome P450
[CYP] enzymes) or reductive (mediated by aldoketoreductases) (
10,
11). Phase II reactions increase the water solubility of a drug or its phase I metabolite to improve the body’s ability to excrete the compound; these reactions conjugate a drug with moieties such as glucuronic acid. Microsomal epoxide hydrolase and uridine diphosphate (UDP)-glucuronosyltransferases are examples of phase II enzymes (
10,
11). Phase I reactions are considered bioactivating; phase II reactions are considered a form of detoxification (
10).
Genetic variability in phase I metabolizing enzymes can alter pharmacokinetic profiles and subsequently drug toxicity. The hepatic microsomal CYP enzymes, the best-studied examples, are coded by a superfamily of CYP genes and play a key role in the metabolism of phenytoin, phenobarbital, carbamazepine, diazepam, ethosuximide, zonisamide, felbamate, and tiagabine (
Table 52.1) (
12,
13). Not all AEDs are metabolized by this enzyme system. Some either do not undergo metabolism (gabapentin, levetiracetam) or are metabolized through alternative non-CYP pathways (lamotrigine by glucuronidation, valproic acid by β-oxidation and glucuronidation, and oxcarbazepine by aldoketoreductases).
Cytochrome P450 Genes
The CYP enzymes metabolize compounds by catalyzing the insertion of an oxygen atom from O2 into an aromatic or aliphatic molecule to form a hydroxyl group. CYP enzymes are heme-thiolate proteins found in all living organisms (
14). In mammals, for example, they are membrane bound and concentrated in the liver. The term
P450 comes from the observation that a broad-band peak occurs at a wave length of 450 nm when a difference spectrum is plotted between reduced CYP treated with nitrogen and reduced CYP treated with carbon monoxide placed in the path of a double-beam spectrometer (
15).
Sequence similarities have been used to devise a standardized nomenclature for categorizing the P450 proteins into families and subfamilies (
14,
16). P450 proteins are in the same family if they exhibit more than 40% similarity in protein sequence; within the same family, proteins that have more than 55% sequence homology are in the same subfamily (
16). Families are given a unique Arabic number; subfamilies noted by a letter after the family number and individual genes in the subfamily are denoted by a second Arabic number after the subfamily letter, for example, CYP2C9 (
7,
14,
16,
17). The website http://www.imm.ki.se/cypalleles is the most informative source for data about CYP allelic variants.
In humans, 59 CYP isoenzymes have been individualized to date and distributed in 42 subfamilies and 18 CYP families. CYP2C9, CYP2C19, and CYP3A4 are particularly important to AED interactions; CYP2C9 and CYP2C19 display noteworthy pharmacogenetic polymorphisms (
18). The CYP2C subfamily accounts for approximately 18% of the hepatic CYP content in humans (
19).
CYP2C9
CYP2C9 is the principal CYP2C isoenzyme in the human liver (
20). The CYP2C9 gene, mapped to chromosome 10q24.2, encompasses nine exons and codes for a protein of 490 amino acids (
19,
21). Of the 13 CYP2C9 alleles identified, CYP2C9
*1, the most common, is considered the wild-type allele (
19,
22). Individuals homozygous for this allele are called extensive metabolizers. Four variant alleles, CYP2C9
*2, CYP2C9
*3, CYP2C9
*4, CYP2C9
*5, have been associated with significant reductions in the metabolism of CYP2C9 substrates, compared with the wild-type allele (
23,
24,
25,
26). CYP2C9
*6 has been linked to inactive CYP2C9 activity (
27). Individuals with at least one of these mentioned variant alleles are called poor metabolizers.
While two-thirds of whites possess the wild-type allele, one-third are heterozygous for CYP2C9
*2 or CYP2C9
*3 (
19). CYP2C9
*2 and CYP2C9
*3 are much less prevalent in African Americans and Asians, more than 95% of whom express the wild-type genotype (
19). To date, CYP2C9
*4 has been identified exclusively in Japanese and CYP2C9
*5 and CYP2C9
*6 in African American populations (
26,
27,
28). Six of the latest seven alleles (CYP2C9
*7 through CYP2C9
*12) were discovered by resequencing CYP2C9 DNA from three racial groups, whites, Asians, and Africans (African Americans and African Pygmies) (
29). CYP2C9
*13, the last discovered allele, was identified in the Chinese, and found to be associated with reduced plasma clearance of drugs that are substrates for CYP2C9 (
30).
CYP2C19
Mapped to chromosome 10q24.1-q24.3, the CYP2C19 gene consists of 9 exons encoding a protein of 490 amino acids (
18). There are 15 alleles, including the wild-type CYP2C19
*1 (
22). The first seven variants (CYP2C19
*2 to CYP2C19
*8) are inactive mutations responsible for the poor-metabolizer phenotype. The recently described CYP2C19
*9 to CYP2C19
*15 are potentially defective, although none has yet been studied
in vivo (
22,
31).
Most individuals in all populations studied have the CYP2C19 extensive-metabolizer phenotype involving the wild-type allele. CYP2C19 poor metabolizers are much more numerous among Asians (13% to 23%) than among whites and African Americans (1% to 6%) (
18). The CYP2C19
*2 and CYP2C19
*3 mutations are responsible for the majority of CYP2C19 poor metabolizers. The main defective allele, CYP2C19
*2, occurs in 30% of Chinese, approximately 15% of whites, and approximately 17% of African Americans. CYP2C19
*3 affects approximately 5% of the Chinese, and is almost non-existent in Caucasians (
32). Both alleles together can explain all Asian and approximately 80% of whites poor metabolizers (
33).
CYP3A4 and CYP3A5
CYP3A4 and CYP3A5 appear to metabolize many of the same drugs, and an exclusive substrate for either has yet to be identified. The most abundant form of CYP in the human liver, CYP3A isoenzymes account for approximately 30% of the total CYP protein (
34) and metabolize about half of all prescribed drugs (
35). The CYP3A locus is found on 7q22.1 and consists of four members; CYP3A4 and CYP3A5 are the main isoenzymes in the liver (
34,
36). Each CYP3A gene contains 13 exons and encodes a 503-amino-acid protein (
37).
Most of the 39 CYP3A4 variants consist of SNPs (
22,
38); large interethnic differences characterize the distribution of these alleles. CYP3A4
*1B, which is not associated with altered catalytic activity, is present in 9% of whites and 53% of Africans and is absent in the Taiwanese population (
39). CYP3A4
*2 occurs in 2.7% of the Finnish population but is absent in whites from Middle and Western Europe, the Chinese, and blacks (
40,
41).
In vitro, this variant showed a lower intrinsic clearance for nifedipine than the wild-type but was not significantly different for testosterone 6β-hydroxylation. CYP3A4
*3 was first identified in a single Chinese individual and later found in Dutch whites with a frequency of 2.2% (
40,
42). The allelic variants CYP3A4
*4, CYP3A4
*5, and CYP3A4
*6 were found in the Chinese at a frequencies of 3%, 2%, and 1% respectively (
43). Measurement of the ratio of morning spot urinary 6β-hydroxycortisol to free cortisol in persons with these mutations suggests that these alleles may have a decreased activity compared with the wild type. The CYP3A4
*7 through CYP3A4
*13 allelic variants have been described in Middle and Western Europeans; CYP3A4
*12 showed altered catalytic activity for testosterone and midazolam (
41).
CYP3A4
*14, CYP3A4
*15, and CYP3A4
*16 have been described in a study of nine different ethnic populations (
38): CYP3A4
*14 in a single person of unknown ancestry, CYP3A4
*15 in 2% of African Americans, and CYP3A4
*16 in 5% of Mexicans and Japanese. The CYP3A4
*17 variant occurred in 27% of whites, whereas the CYP3A4
*18 and CYP3A4
*19 mutations were observed in Asians at allelic frequencies of 2% (
44).
In vitro assessments of the catalytic activity of CYP3A4 using testosterone and the insecticide chlorpyrifos demonstrated decreased activity for CYP3A4
*17 and increased activity for CYP3A4
*18. Overall, the heterozygous frequency of at least one nonsynonymous CYP3A4 mutant allele was 14% in whites, 10% in Japanese, and 15% in African Americans and Mexicans (
38).
CYP3A5 is one of the two most important CYP3A proteins in the liver (
45). Expression is variable, with reported rates in 10% to 29% of livers to at least trace presence in all liver samples, depending on the detection method used (
46,
47,
48,
49). CYP3A5 is more frequently expressed in hepatic samples of African Americans than in those from whites (60% versus 33%) (
36).
Twenty-three variants of CYP3A5 have been described to date (
22); however, only individuals with the wild-type CYP3A5
*1 allele produce high levels of full-length CYP3A5 messenger RNA and express the CYP3A5 protein (
36). CYP3A5
*1 occurs at the following frequencies: 15% in whites and Japanese, 25% in Mexicans and Southeast Asians, 33% in Pacific Islanders, 35% in Chinese, 45% in African Americans, and 60% in Southwestern American Indians (
36). The CYP3A5
*2 variant was described in two of five whites with no CYP3A5 protein (
50) but was not found in the Chinese or African-American populations (
45,
51). The presence of CYP3A5
*3 is the most common cause of the absence of the CYP3A5 isoenzyme, which results from alternate splicing and truncation of the protein (
36). The frequency of this mutation varies from 27% in African Americans to 75% in Asians and 95% in whites (
37,
52). CYP3A5
*4 and CYP3A5
*5 were each found in 1.8% of Chinese persons (
51). CYP3A5
*6 was identified in 15% of African Americans and was associated with either normal or decreased enzyme activity (
36). Ten percent of African Americans had the CYP3A5
*7 mutation; correlation with enzymatic activity was unclear (
45). The CYP3A5
*8 variant occurs in African populations with a frequency of 4%; CYP3A5
*9 is present in 2% of Asians; and CYP3A5
*10 is found in 2% of whites (
52). These three mutations exhibited decreased enzymatic activity for testosterone clearance and nifedipine oxidation, compared with the wild-type allele (
52).