aAfter administration of eslicarbazepine acetate.
CYP, Cytochrome P450; UGT, UDP-glucuronosyltransferase; NAT, N-acetyltransferase; nk, not known.
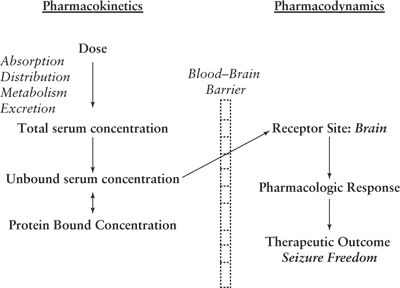
Figure 44.1. Relationship between pharmacokinetics and pharmacodynamics.
PHARMACOKINETIC PARAMETERS
Absorption
Absorption refers to the passage of the drug from its site of administration into the systemic circulation and is defined by the rate at which the drug leaves the site of administration and the extent at which it occurs. The rate of absorption is generally a first-order process, where the rate of absorption is dependent on the amount of drug. Some drugs can follow zero-order kinetics with a constant release of drug independent of the amount of drug. Bioavailability (F) is the amount of the administered drug that reaches the systemic circulation. F is dependent on the fraction absorbed through the gastrointestinal (GI) tract plus the fraction lost due to first-pass metabolism. After intravenous (IV) administration, bioavailability is equal to 1.0. Bioavailability for other non-IV formulations is determined by comparing the area under the concentration time curve (AUC) obtained after IV administration of a drug and the AUC obtained after administration by another route (i.e., oral). Most drugs are absorbed by passive diffusion in the GI tract where the rate of absorption is proportional to the drug concentration gradient across the barrier. Other drugs are absorbed by a combination of passive and active transport by proteins that can increase and/or decrease absorption depending on their location and whether they are influx or efflux transporters (1). Influx transporters that increase oral absorption include the organic anion transporters (OAT), organic cation transporters (OCT), organic anion–transporting polypeptides (OATP), peptide transporters, and large neutral amino acid transporter 1 (LAT). The oral absorption of gabapentin is dependent on active transport by the LAT (2). Conversely, efflux transporters, including P-glycoprotein (PGP), breast cancer resistance protein (BCRP), multidrug resistance proteins (MRP), and multidrug and toxin extrusion proteins (MATE), can limit drug absorption by increasing the excretion of drugs into the intestinal lumen from the systemic circulation (3). First-pass metabolism of a drug can occur in the GI tract and in the liver resulting in a decreased bioavailability. The fraction lost in the GI tract can be due to metabolism via hydrolysis, glucuronidation, sulfation, and oxidation, with cytochrome P450 3A4 (CYP3A4), the most dominant cytochrome P450 in the GI tract. Many drugs, including carbamazepine (4), are substrates of PGP and CYP3A4, both involved in decreasing oral absorption. Drugs that are high extraction ratio (ER) drugs can undergo significant first-pass liver metabolism.
Extended-release formulations are used to decrease the frequency of dosing for drugs with rapid elimination to improve convenience and compliance. For extended-release drugs, the rate-limiting step in drug elimination is the absorption rate of the drug and not the elimination rate. Use of an extended- release product can decrease the peak to trough fluctuation in serum concentrations and theoretically improve the therapeutic benefit of the drug by decreasing adverse events associated with higher peak concentrations. Other drugs are delayed release, for example, enteric-coated valproate. The enteric coating improves tolerability by decreasing absorption within the stomach and delaying absorption until the formulation reaches the intestines.
Bioequivalence is defined as chemical, when the drug meets the same chemical and physical standards; biologic, when the administered drug yields similar concentrations in blood; and therapeutic, when the drug provides equal therapeutic benefits in clinical trials. Generic drugs are chemically and biologically equivalent. Manufacturers do not have to prove therapeutic equivalence. Bioequivalence studies are single-dose, crossover studies done in healthy subjects comparing a generic product to the brand-name product. A generic product is considered bioequivalent if the mean AUC, maximum concentration (Cmax), and the 90% confidence interval for each parameter are within 80% and 125% of the brand product. There has been a history of problems with generic versions of the older AEDs, specifically carbamazepine and phenytoin, as summarized in several reviews. Nuwer et al. (5) described three pharmacokinetic properties that predisposed the older AEDs to problems with their generic formulations: low water solubility, narrow therapeutic range, and nonlinear pharmacokinetics. The Biopharmaceutics Classification System (BCS) was designed to correlate a drug’s aqueous solubility and permeability with the rate and extent of oral drug absorption (6). BCS can also provide an estimate for the likelihood of problems with generics. Drugs with high solubility and permeability are unlikely to demonstrate generic-related problems. Using the criteria, the historical problems with generic products of phenytoin and carbamazepine are predictable. The same criterion predicts that the majority of the new AEDs should not be predisposed to similar problems due to their high aqueous solubility and permeability. There have been reports from patients and neurologists of increased side effects or seizures on generic substitution (7). A recent evaluation of 141 generic AED products in 258 bioequivalence studies found that there was less than a 15% difference in the AUC and Cmax compared to the reference product in 99% and 89% of the studies, respectively (8). The AED with the lowest solubility, oxcarbazepine, had the highest variability. In a systematic review of published 13 prospective and 7 retrospective studies, the highest quality studies (prospective, class I and II evidence), found that there was no significant difference between brand and generic formulations in safety and efficacy (9). In contrast, several retrospective studies of lower quality (class III evidence) did find a difference.
Distribution
Distribution is the process of reversible transfer of drug to and from the site of measurement. Central nervous system distribution is unique due to the blood–brain barrier (BBB). Lipid-soluble and unbound drugs have significantly higher distribution across the BBB than do water-soluble and protein-bound drugs. Both influx and efflux transport proteins alter brain distribution (3). Influx transporters involved in drug distribution include LAT, monocarboxylate transporter 1 (MCT1), and OATP. The brain uptake of gabapentin and pregabalin is dependent on the influx transporter, LAT1 (10). PGP and BRRP are the primary efflux transporters involved in transporting drugs from the brain to the blood and form an important part of the BBB. After initial reports of the overexpression of PGP in patients with refractory epilepsy (11), it was hypothesized that increased expression of drug efflux transporters, specifically PGP and MRPs, reduce the concentration of AEDs in the brain resulting in pharmacoresistance (12). Although there is experimental evidence that phenytoin, phenobarbital, lamotrigine, and oxcarbazepine are PGP substrates, many of the other AEDs that are ineffective in refractory epilepsy are not substrates, suggesting there is not a major role of efflux transporters in pharmacoresistance (13,14).
The volume of distribution (Vd) is a measure of the apparent space in the body available to contain the drug. Vd relates the amount of drug in the body to the concentration of drug in the plasma. Therefore, the initial concentration (Co) attained after administration of a single or bolus dose (D) is dependent on the Vd of the drug. The dose is based on either ideal or total body weight depending on the physiochemical characteristic of the drug. Lipophilic drugs will distribute into adipose tissue, and Vd will be dependent on total body weight. In contrast, for water-soluble drugs, Vd is dependent on ideal or lean body weight. Vd can be used to calculate both loading and bolus doses needed to achieve a desired concentration.
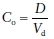 (1)
(1)For the AEDs, albumin is the primary binding protein, with the exception of carbamazepine, which is bound to both albumin and alpha1-acid glycoprotein (α1-AGP). Albumin concentrations are decreased in the neonate, in the elderly, in hepatic and renal disease, during pregnancy, and after trauma. α1-AGP is an acute-phase reactive protein and is decreased in neonates and increased in conditions of inflammation and trauma. For the majority of the drugs, protein binding is linear, and the percent unbound is a constant within the range of concentrations used clinically. Valproate is the one exception. Valproate is highly protein bound, and due to its high molar concentration, valproate saturates albumin-binding sites within the therapeutic range (15). An increase in the percent unbound as the dose increases results in measured total valproate concentrations increasing less than proportionally with increasing doses. Conversely, unbound valproate concentrations will increase linearly with increasing dose, and total valproate concentrations will no longer reflect unbound or active concentrations.
Elimination Processes
Drugs are eliminated by metabolism and/or excretion of unchanged drug by the kidneys or GI tract. Metabolism occurs predominantly in the liver with the GI, kidneys, lung and serum as other possible sites of metabolism. For the large majority of drugs, elimination is linear; the elimination rate is proportional to the amount of drug present. For drugs following linear kinetics, clearance is constant, and serum concentrations increase proportional with increasing doses. Unlike other drugs, phenytoin is unique in that its elimination is nonlinear due to saturation of metabolism within the normal dosage range. This saturation of metabolic processes results in a decreased clearance with increasing doses. For drugs like phenytoin with nonlinear elimination, serum concentrations will increase more than expected with increasing doses.
Clearance (Cl) is the most useful pharmacokinetic parameter for evaluating an elimination mechanism. It can also be used to estimate the average steady-state concentrations (Cave,ss) attained on multiple dosing. Physiologically, Cl is the loss of drug across an organ of elimination and is determined by the blood flow to the organ that metabolizes or eliminates the drug and the efficiency of the organ in extracting the drug. The efficiency is measured by the ER, defined as the ratio of the difference between the concentration into and out of the organ (Cin – Cout) to the concentration entering the organ (Cin). Cl is described in terms of the eliminating organ; hepatic clearance (ClH) and renal clearance (ClR) with total Cl determined by the sum of all the partial clearances. After multiple dosing, Cave,ss is dependent on the dose/interval (D/τ), Cl, and F (Eq. (2)).
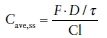 (2)
(2)The elimination half-life (T1/2) is the time required for the serum concentrations to decrease by 50% and is independent of dose. As shown in Eq. (3), T1/2 is dependent on the Cl and Vd. It takes five T1/2s to reach steady-state concentrations with multiple dosing and to eliminate greater than 95% of the drug on discontinuation.
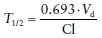 (3)
(3)Excretion
The renal excretion of unchanged drug and metabolites includes the processes of glomerular filtration (GFR), active tubular secretion, and passive reabsorption. The major transporter involved in renal secretion of drugs are OAT, OCT, PGP, and MRP (3). For drugs that are predominantly excreted unchanged in the urine, renal function can be assessed using the serum creatinine (SCR) to estimate creatinine clearance (CrCl) or GFR (eGRF). The Cl of creatinine is dependent on GFR and tubular secretion and is greater than GFR. The relationship between a drug’s Cl and CrCl (or eGFR) can be used to estimate doses regiment needed to attain therapeutic concentrations. For most drugs that are significantly excreted unchanged in the urine, the relationship between Cl and CrCl (or eGFR) is linear with the y-intercept reflecting the nonrenal portion of the Cl. CrCl can be estimated using the Cockroft–Gault equation (16), the modified diet in renal disease (MDRD) equation (17), or the chronic kidney disease epidemiology (CKD-EPI) (18). The Cockroft–Gault equation estimates CrCl and requires knowledge of the patients’ age, sex, and lean body weight (LBW). The MDRD equation was developed in 1070 patients with chronic renal failure. More recently, the CKD-EPI method was developed using a cross-sectional analysis with pooled data obtained from research studies and clinical population of 8254 patients with measured GFR. The CKD-EPI method performs better at higher GFR. The MDRD and CKD-EPI equations estimate GFR, do not require information on LBW, and also take into account ethnic differences in renal function. The majority of clinical laboratories in the United States were reporting eGFR with a measured SCR using the MDRD equation in 2008. More recently, the CKD-EPI is now used by QUEST and LabCorp, the two largest laboratory service providers in the United States (19). For the majority of the currently marketed drugs, the Cockcroft–Gault method was used to estimate renal function and to determine the relationship between CrCl and Cl (or Cl/F) to recommend dosing guidelines. As clinical laboratories are reporting eGFR, Park et al. evaluated data in the FDA database for 26 approved drugs that require renal dosage adjustment to determine if the MDRD equation could be used interchangeable with the Cockcroft–Gault (20). Only 8% of the dosage recommendations were changed to a different dosage class if the MDRD determined eGFR adjusted for BSA (20). Therefore, the eGFR can be used to guide drug dosing.
Excretion of drugs into breast milk can occur during lactation. The dose of a drug that the infant receives during breast-feeding is dependent on the amount excreted into the breast milk, the daily volume of milk ingested, and the average plasma concentrations of the mother. The physiochemical properties of a drug will determine how much of the drug will be excreted into the breast milk, including its lipophilicity and protein-binding and ionization properties. The milk-to-plasma concentration ratio has large inter- and intrasubject variability and is often not known. In contrast, protein binding is usually known, and knowledge of the protein-binding properties of a drug can provide a quick and easy tool to estimate exposure of an infant to medication from breast-feeding. Based on an extensive literature review of case reports that included infant concentrations from breast-fed infants exposed to maternal drugs (21), measurable concentrations of drug in the infant did not occur for drugs that were at least 85% protein bound, if there was no placental exposure immediately prior to or during delivery.
Metabolism
Metabolic reactions are primarily catalyzed by the cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) enzymes. However, the second- and third-generation AEDs are also metabolized by a variety of other non-CYP/UGT enzymes (Table 44.1). CYPs are a family of multiple enzymes with the individual isozymes being composed of three major families (CYP1, CYP2, and CYP3). Seven primary isozymes are involved in the hepatic metabolism of most drugs: CYP1A2, CYP2A6, CYP2C8/9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4. The most abundant isozyme, CYP3A4, which accounts for approximately 30% of the total hepatic CYP, has the broadest substrate specificity and is involved in the metabolism of more than 50% of all drugs. The UGTs are family of enzymes that catalyze the transfer of a glucuronic acid moiety from a donor cosubstrate UDPGA. The activity of the metabolic enzymes is dependent on genetic, physiologic, and environmental effects.
Genetic polymorphisms in the expression of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A5, UGT1A1, N-acetyltransferases (NAT2), and thiopurine S-methyltransferase (TPMT) have been identified. Poor metabolizers are homozygous for the mutant gene. Extensive metabolizers are either homozygous or heterozygous for the wild-type gene, with heterozygous carriers having intermediate metabolic activity. Ultrametabolizers have multiple copies of the gene for CYP2D6 and a promoter region polymorphism for CYP2C19 resulting in increased metabolism of CYP2D6 and CYP2C19 substrates, respectively. In addition, the predominant variants of CYP2D6 in Asian and African-American (*10 and *17, respectively) are alleles with reduced enzyme activity. Overall, there is a large interethnic variability in the proportion of poor metabolizers, intermediate metabolizers, and ultrametabolizers, which is beyond the scope of this chapter (22).
The pharmacokinetic hepatic model of elimination is a physiologically based model where hepatic clearance (ClH) is dependent on the unbound fraction of the drug in the blood (fu), activity of the metabolic enzymes (Clint), and hepatic blood flow (QH).
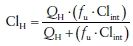 (4)
(4)For low extraction ratio drugs (ER < 0.3), and fu · Clint < QH, Cl is dependent on protein binding and intrinsic clearance. For high ER drugs (ER > 0.7), fu · Clint > QH and Cl is dependent on QH.
Protein Binding and Hepatic Metabolism
Protein-binding effects are only clinically significant for two different types of highly protein-bound drugs that are predominantly eliminated by hepatic elimination (23). For low ER drugs that undergo dosage adjustment by monitoring total concentrations, total concentrations will underestimate unbound or active concentrations. This has been shown for both phenytoin and valproate in the elderly (24) and in pregnant women (25,26) with epilepsy. Total concentrations decreased significantly more than unbound concentrations with decreased albumin concentrations. Adjusting doses based on total concentrations will result in higher doses of valproate and phenytoin than needed to maintain therapeutic unbound concentrations. As the teratogenicity of valproate and phenytoin has been found to be dose dependent, minimizing unnecessary dosing increases during pregnancy is desirable. Ideally, unbound concentrations should be measured. Unbound phenytoin concentrations are clinically available and should be utilized whenever possible. Valproate unbound concentrations are not routinely available to the clinician as they may be unreliable due to problems in sample collection. Patients need to be monitored based on clinical measures, that is, a change in seizure frequency and/or presence of adverse effects in the case of the AEDs.
For high ER drugs with a narrow therapeutic window and administered by nonoral routes, the AUC of unbound drug can be significantly increased and result in increased pharmacologic effect. The AUC of low ER drugs is independent of protein binding (23). Of the parenterally available AEDs, only midazolam is a high protein bound, and high ER drug.
METHODS TO DETERMINE PHARMACOKINETIC PARAMETERS
Pharmacokinetic parameters are determined using concentration time data by both compartmental and noncompartmental methods of analysis. The majority of drugs are eliminated by a first-order process, that is, the elimination rate is dependent on concentration, and serum concentration time data can be modeled using either a one-compartment or a two-compartment model. For one-compartment model, distribution is considered instantaneous, and concentrations decline exponentially with time, and a plot of log concentration versus time is linear. For a two-compartment model, the first exponential phase is primarily dependent on distribution into a peripheral compartment, and the second exponential phase is dependent on elimination after distribution is complete. The terminal elimination rate constant (β) is determined by linear regression of the log concentration time data obtained during the terminal exponential phase. The elimination half-life (T1/2) is calculated as 0.693/β. Peak serum concentration (Cmax) and the time to peak (Tmax) are obtained by visual inspection of the data. The AUC is calculated by the log-linear trapezoidal method. Cl is estimated by the ratio of dose to AUC. If the drug is administered orally, an apparent oral clearance (Cl/F) is defined as D/AUC as bioavailability is unknown or assumed. Renal clearance (ClR) is calculated as product as the fraction of the dose excreted unchanged in the urine and Cl.
PHYSIOLOGIC AND PATHOLOGIC EFFECTS ON PHARMACOKINETICS
The effect of age (27,28), pregnancy (21,29), drug interactions (30–32), renal disease (33), and/or hepatic disease (34) on AED dosing will depend on the fraction of the AED eliminated by renal and/or hepatic elimination, the metabolic isozymes involved, as well as the extent of protein binding, if therapeutic drug monitoring is involved (Table 44.1). This section provides the theoretical basis for the physiologic and pathologic effects on drug pharmacokinetics. Refer to review articles (9,13–19) and chapters on individual AEDs for more specific information.
Age
Gastric pH is increased in neonates, infants, and young children, decreasing to adult values by 2 years of age. The incidence of hypochlorhydria also increases significantly after age 70. GI motility is decreased in neonates, reaches adult levels in older infants, and then decreases in the elderly. The bioavailability of drugs given orally that are weak acids, like phenytoin and phenobarbital, may be decreased in infants, young children, and the elderly due to their higher gastric pH. In neonates and infants, the increased total body water-to-body fat ratio contributes to an increase in the Vd of hydrophilic drugs, and may require larger mg/kg loading doses of some drugs to achieve therapeutic concentrations. Albumin and α1-AGP concentrations are decreased in the neonates and young infant. Albumin concentrations alone are decreased in the elderly resulting in decreased protein binding for highly bound drugs. The clinical significance of decreased protein binding is described above for low and high ER drugs. At birth, GFR is approximately 40 mL/min/1.73 m2 in the full-term neonate and increases steadily to 80% to 90% of adults function by 1 year. From ages 40 to 80, kidney function declines at approximately 10% to 20% decrease per decade. Therefore, in general, weight-normalized doses of drugs excreted predominately unchanged by the kidneys need to be reduced in neonates, infants, and the elderly. In the past, there was a general assumption that all hepatic drug metabolism was increased in children compared to adults. There is now evidence that in children, hepatic metabolism can be increased or not changed compared to adults depending on metabolic pathway involved in the elimination of the drug (26). Significantly, higher weight-corrected doses are needed in children than in adults for drugs metabolized by CYP1A2, CYP2C9, and CYP3A4. In contrast, weight-corrected doses for drugs metabolized by CYP2C19, CYP2D6, NAT2, and UGT in children are similar to those in adults. In the elderly, the activity of all of the CYPs is decreased resulting in a need for decreased doses. Doses of drugs metabolized by the conjugating enzymes, NAT2 and UGT, do not need to be decreased.
Pregnancy
Despite the many physiologic changes that occur during pregnancy that could affect absorption, bioavailability does not appear to be altered. Decreased albumin and α1-AGP concentrations during pregnancy will result in decreased protein binding for highly bound drugs. The clinical significance of decreased protein binding is described above and is important for both valproate and phenytoin, specifically. Renal clearance and the activity of the CYP3A4, CYP2D6, CYP2C9, and UGT isozymes are increased during pregnancy, and drugs eliminated by these pathways may need dosage increases during pregnancy (25,26). In contrast, CYP1A2 and CYP2C19 activity is decreased, and drugs metabolized by these CYP isozymes may require a decrease in dosage during pregnancy (21).
Renal Disease
The effect of renal disease, in addition to declining renal function in the elderly and the immature renal function in neonates and infants, all result in decreased Cl for drugs that are predominantly excreted unchanged in the urine. For drugs with both renal and hepatic Cl, the effect of renal dysfunction is proportionate to the fraction of the Cl of the drug dependent on renal clearance. The ability to estimate renal function using SCR provides a method of determining the dose required to maintain a therapeutic effect without toxicity. In addition, to a decreased Cl, renal disease is associated with decreased albumin concentrations, which will result in decreased protein binding for highly bound drugs.
Liver Disease
Unlike renal disease, there are no specific markers of liver function that can be used to provide guidance in dosage adjustments. Liver function tests (LFTs) primarily are a measure of the extent of cell death. For example, LFTs are significantly increased in acute hepatitis; however, metabolic enzyme function is maintained, and doses of drugs eliminated by liver metabolism do not need to be altered (26,27). Conversely, acute and chronic cirrhosis results in a decreased ability to metabolize drugs due to effects on the metabolic enzymes and hepatic blood flow. The metabolic enzymes are differentially affected depending on the severity of the cirrhosis with the Cl of drugs metabolized by CYP2C19 affected first during mild liver disease (34). The activity of CYP2C19 is significantly decreased with mild liver disease and remains at a decreased level with increasing severity of disease. CYP1A2, CYP2D6, and CYP3A4 activity is also decreased in mild cirrhosis, but to a lesser extent than CYP2C19, and continues to decrease with increasing severity of disease. CYP2C9 and CYP2E1 are not significantly decreased in mild to moderate liver disease; however, the activity of the enzymes all decrease with more severe cirrhosis. The Cl of drugs metabolized by the conjugating enzymes (UGT and NAT2) is not affected until the patient reaches end-stage liver disease. End-stage liver disease also decreases the renal clearance of drugs. Decreased albumin and α1-AGP concentrations occur due to decreased synthesis and result in increased free fractions of highly protein-bound drugs.
DRUG INTERACTIONS
Drug interactions can occur by pharmacokinetic or pharmacodynamic mechanisms. Pharmacodynamic interactions occur when the pharmacology of one agent alters the pharmacology or effect of the other drug without altering the serum concentration. Theoretically, the interaction can occur at the receptor or site of action or indirectly by affecting other physiologic mechanisms. The most commonly occurring AEDs are pharmacokinetic interactions, where one drug alters the serum concentrations of another. Pharmacokinetic interactions include hepatic enzyme induction, enzyme inhibition, and protein-binding displacement. Many of the AEDs are either specific or broad-spectrum inducers and/or inhibitors of metabolic enzymes (Table 44.2). In addition, many of the AEDs are eliminated by pathways that are affected by induction and/or inhibition by other AEDs and non-AEDs (Table 44.3).
Table 44.2 Effect of AEDs on Hepatic Metabolic Enzymes
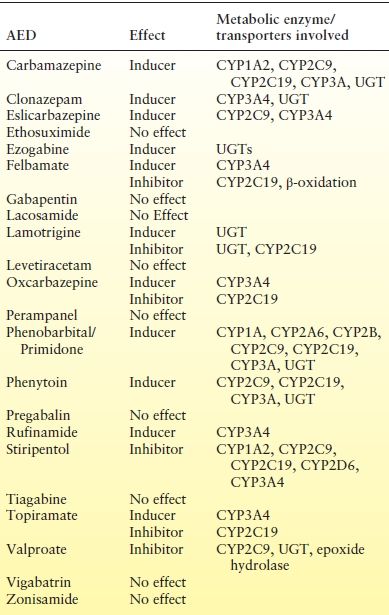
CYP, Cytochrome P450; UGT, UDP-glucuronosyltransferase; NAT, N-acetyltransferase; PG, p-glycoprotein; MRP, multiresistance protein.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


