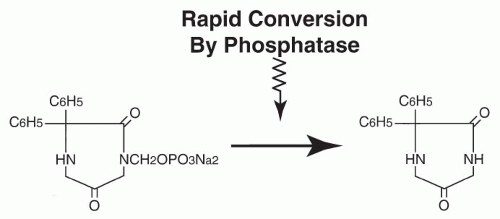
Absorption
Phenytoin is available in various formulations for both oral and parenteral use (
Table 55.1). Both the rate and extent of absorption may differ among the formulations, leading to clinically significant alterations in serum concentrations when switching among products.
The rate and extent of absorption of phenytoin from its site of entrance depends on pKa and lipid solubility, the pH of the medium in which it is dissolved, solubility in the medium, and concentration. These factors are frequently altered by the presence of foods or drugs in the intestinal tract and by the formulations. Little phenytoin is absorbed in the stomach because the drug is insoluble in the acidic pH of gastric juice (about 2.0), even though it is in its nonionized form in the stomach. Absorption occurs primarily in the duodenum, where the higher pH increases the solubility of phenytoin. Absorption from the jejunum and ileum is slower than from the duodenum and is poor from the colon (
17,
18).
In humans, the rate of absorption is variable and prolonged (
19,
20), and significantly influenced by the rate of elimination (
21). Because dissolution is the rate-limiting process in the absorption of phenytoin, any factor that affects dissolution or solubility will affect absorption. After oral administration of a single dose, peak blood drug levels are generally reached between 4 and 8 hours later (range, 3 to 12 hours) (
22,
23). In patients ingesting massive amounts of phenytoin, absorption may continue for as long as 60 hours (
24). Relative bioavailability increases with age, suggesting an age-dependent effect on drug absorption (
25). In newborns and infants up to 3 months old, phenytoin is absorbed slowly and incompletely after both oral and
intramuscular administration (
26); absorption in older infants and children is similar to that in adults. Stable isotope tracer doses have been used to assess the bioavailability of phenytoin (
27,
28).
After intramuscular administration, phenytoin is absorbed slowly, as poor water solubility leads to precipitation of drug at the injection site, forming almost a depot repository (
20). This prolonged absorption and pain on administration mandate use of the intravenous route if parenteral administration is required.
The reported bioavailability of rectally administered phenytoin sodium is approximately 25% (
29).
Metabolism
In humans, the major pathway of phenytoin elimination (approximately 80%) is 4′- hydroxylation to form 5-(4′-hydroxyphenyl)-5-phenylhydantoin (4′-HPPH). This reaction is mediated mainly by the cytochrome P450
(CYP) enzyme CYP2C9, and to a lesser extent by CYP2C19 (
47,
48). Approximately 10% of phenytoin is eliminated to a dihydrodiol, and another 10% is metabolized to 5-(3-hydroxyphenyl)-5-phenylhydantoin (3′,4′-diHPPH) (
7,
47,
49). An arene oxide, which precedes the formation of these compounds, has been implicated in the toxicity and teratogenicity of phenytoin; however, its transient presence in patients with normally functioning arene oxide detoxification systems is unlikely to account for many of the toxic reactions (
50,
51).
Because phenytoin has nonlinear pharmacokinetics, a narrow therapeutic index, and a concentration-related toxicity profile, small changes in CYP2C9 activity may be clinically significant. Of the 13 CYP2C9 alleles identified to date, the most common, designated as CYP2C9
*1, is considered the wild-type allele (
52,
53). Individuals homozygous for the wild-type allele are called extensive metabolizers. Studies in various populations demonstrated that the CYP2C9
*2, CYP2C9
*3, CYP2C9
*4, and CYP2C9
*6 alleles are important
in vivo determinants of phenytoin disposition (
54,
55,
56,
57,
58,
59,
60,
61,
62,
63). Individuals with at least one of these variant alleles are called poor metabolizers and have a reduced ability to metabolize phenytoin. They may require lower-than-average phenytoin doses to decrease the incidence of concentration-dependent adverse effects (
58,
64).
Although two-thirds of whites possess the wild-type allele, one-third are heterozygous for the CYP2C9
*2 or CYP2C9
*3 allele (
52). These two variant alleles are much less prevalent in African Americans and Asians, with more than 95% of these groups expressing the wild-type genotype (
52). To date, the CYP2C9
*4, CYP2C9
*5, and CYP2C9
*6 allelic variants have been identified exclusively in the Japanese (CYP2C9
*4) and African-American (CYP2C9
*5 and CYP2C9
*6) populations (
63,
65,
66). Six of the latest seven alleles (CYP2C9
*7 through CYP2C9
*12) have been discovered by resequencing CYP2C9 DNA from whites, Asians, and Africans (African Americans and African Pygmies) (
67). CYP2C9
*13 was identified in a Chinese population, and found to be associated with reduced plasma clearance of drugs that are substrates for CYP2C9 (
68).
Odani and coworkers observed a decrease of approximately 30% in the maximal rate of phenytoin elimination in Japanese heterozygous for CYP2C9
*3 compared with those homozygous for the wild-type allele (
54). Moreover, the mean phenytoin maintenance dose leading to a therapeutic serum concentration was significantly lower in patients with CYP2C9 allelic variants (199 ± 42.5 mg/day) than in those with the wild-type allele (314 ± 61.2 mg/day;
p <0.01) (
58). A case report of a heterozygous CYP2C9
*3 allele carrier described excessive phenytoin concentrations relative to the doses taken; a toxic level (32.6 μg/mL) was reached despite a modest dose (187.5 mg/day). The patient showed signs of central nervous system intoxication, ataxia, and diplopia (
59).
The activity of CYP2C9 alone, however, does not fully explain the large interindividual variability in the clinical pharmacokinetics and reported drug interactions of phenytoin (
69). Fifteen CYP2C19 alleles have been described to date (
53). The first seven (CYP2C19
*2 to CYP2C19
*8) are inactive and are responsible for the poor-metabolizer phenotype. The recently described CYP2C19
*9 to CYP2C19
*15 are potentially defective, although none have yet been studied
in vivo (
70).
The majority of all populations studied have the CYP2C19 extensive-metabolizer phenotype involving the wild-type CYP2C19
*1 allele. The frequency of CYP2C19 poor metabolizers is much higher in Asians (13% to 23%) than in whites and African Americans (1% to 6%) (
71). The CYP2C19
*2 and CYP2C19
*3 mutations are responsible for most of the CYP2C19 poor metabolizers. CYP2C19
*2, the main defective allele, occurs with a frequency of 30% in the Chinese population, approximately 15% in whites, and approximately 17% in African Americans. The CYP2C19
*3 variant affects approximately 5% of Chinese, and is almost nonexistent in whites (
72). Together, the CYP2C19
*2 and CYP2C19
*3 alleles can explain all Asian and approximately 80% of white poor metabolizers (
73).
Because the contribution of CYP2C19 to the metabolism of phenytoin increases with an increase in drug concentration, CYP2C19 may be important when CYP2C9 is saturated. The reported differences in K
m values for CYP2C9-catalyzed and CYP2C19-catalyzed phenytoin hydroxylation (5.5 μmol/L versus 71.4 μmol/L) suggest that CYP2C9 is likely to become saturated at phenytoin therapeutic concentrations of 10 to 20 μg/mL (40 to 80 μmol/L) (
74). This mechanism explains the increased risk of toxic reactions with the coadministration of CYP2C19 inhibitors such as ticlopidine or isoniazid. The 1% to 2% of white poor metabolizers for both CYP2C9 and CYP2C19 are particularly susceptible to phenytoin’s adverse effects (
71). Dosage adjustments based on the CYP2C9 and CYP2C19 genotypes may decrease the risk of concentration-dependent adverse effects in allelic variant carriers, particularly at the beginning of therapy.
A Japanese epilepsy study (
54) noted an approximate decrease of 14% in the maximum metabolic rate in patients with CYP2C19 variants compared with those with the extensive-metabolizer phenotype. In another Japanese study (
55), the predicted plasma concentrations with a phenytoin dose of 5 mg/kg per day were 18.7, 22.8, and 28.8 μg/mL in CYP2C19 homozygous extensive metabolizers, heterozygous extensive metabolizers, and poor metabolizers, respectively.
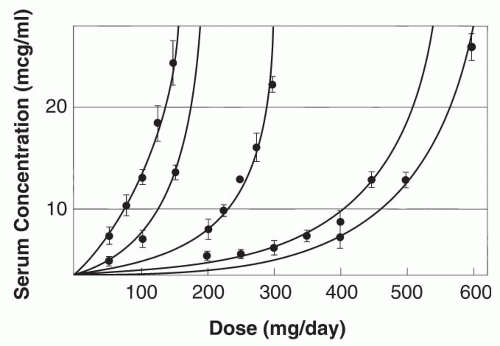
Enzyme saturation kinetics leads to phenytoin plasma concentrations increasing nonproportionally with changes in dose (
Fig. 55.2) (
75). The relationship between dose and concentration can be expressed by the Michaelis-Menten equation:
Dose (mg/day) = VmaxCss/Km + Css
where V
max is the maximal rate of drug metabolism, C
ss the steady-state serum concentration, and K
m the concentration
at which V
max is half-maximal. The mean apparent phenytoin K
m in adults 20 to 39 years old is 5.7 μg/mL (range, 1.5 to 20.7 μg/mL); the mean V
max is 7.5 mg/kg/day (
76). In most patients, phenytoin exhibits nonlinear pharmacokinetics because the usual therapeutic plasma concentrations exceed the usual K
m. Concomitant illnesses (
77) or medications, pregnancy (
78), genetic makeup (
79,
80,
81), and age can significantly affect V
max or K
m (or both). Children have higher V
max values, but similar K
m values, compared with adults (
82,
83,
84); elderly individuals have lower V
max values (mean, 6.0 mg/kg per day) (
76).