Phenytoin, Fosphenytoin, and Other Hydantoins
John M. Stern
Emilio Perucca
Thomas R. Browne
Introduction
The hydantoins are derivatives of a common five-membered heterocyclic ring that differ according to their unique combinations of alkyl and phenyl substitutions at three positions on the ring (Table 1). Phenytoin (PHT) is the most commonly prescribed hydantoin, and fosphenytoin is a phosphate ester prodrug of PHT used for parenteral administration of PHT. Ethotoin and mephenytoin are prescribed less frequently than PHT but may be useful in specific situations.
Phenytoin
Chemistry
The structural formula of PHT (5,5-diphenylhydantoin) is shown in Table 1. PHT has a molecular weight of 252.26 and is a weak acid with a pKa of 8.3 to 9.2 under different experimental conditions.70 Because of its high pKa, PHT is relatively insoluble in water at acid or physiologic pH but is quite soluble in water at alkaline pH. These relationships have important clinical consequences, discussed below. The sodium salt of PHT has a molecular weight of 274.25 and contains the equivalent of 91.98% PHT acid.70
Multiple reliable methods for determining the concentration of PHT in biologic fluids have been reported. These employ gas–liquid chromatography, high-performance liquid chromatography, and immunoassay techniques.87
Pharmacology
PHT has multiple effects on cell function, which presumably explain its therapeutic and toxic effects. Three of these functions seem particularly important. First, PHT causes use-dependent inhibition of sodium channels necessary for the “firing” of action potentials. Sodium channels exist in resting, open, and inactive states. Each time sodium channels open during the passage of an action potential, some sodium channels become inactivated for a period of time before reverting to the resting state. When enough sodium channels become inactivated, the cell can no longer propagate an action potential. By stabilizing the inactivated form of the sodium channel, PHT hastens the process of use-dependent inhibition of action potentials. This phenomenon at least partially accounts for PHT’s ability to control ictal excitability. Second, the ability of PHT to regulate calmodulin and second messenger systems may account for some of its widespread effects on cellular function. Third, PHT has the ability to regulate voltage-dependent neurotransmitter release at the synapse, which may be related in part to its action on calcium or sodium ions. The mechanisms of action of PHT have recently been reviewed in detail elsewhere53 (see Chapter 137).
Clinical Pharmacokinetics
Absorption by Oral Route: General Considerations
As a weak acid with a pKa of approximately 9.0, PHT is <1% ionized and has a water solubility of 14 μg/mL70,189 at a pH of 1 to 2. At a pH of 7.5, PHT is 3% ionized and has a water solubility of 100 μg/mL. Thus, only a small amount of PHT is absorbed in the stomach, and most PHT absorption takes place in the small intestine where its solubility is approximately 100 μg/mL.189
PHT’s low solubility has several important consequences. Not all of an oral dose of PHT is absorbed, and some is lost in the feces. There also are apparently considerable differences in the amount of PHT absorbed by different people, especially when the capsule form is used. Some otherwise normal people require large doses of PHT to reach average drug plasma concentrations, and these people sometimes are accused incorrectly of being noncompliant. Altered PHT absorption is also influenced by certain physiologic states. Pregnancy may be associated with reduced gastrointestinal absorption of PHT.156 Neonates absorb oral PHT incompletely and erratically.16,140 A final consequence of the marginal intestinal solubility of PHT is that any substance that interferes with the dissolution of PHT or adsorbs PHT in intestinal fluids (e.g., nasogastric feedings, antacids, certain foods, and certain other drugs) will inhibit PHT’s absorption.7,189
Differences in Oral Bioavailability among Formulations
There are three types of oral preparations of PHT: Prompt, sustained (extended) release, and liquid suspension. Prompt products are absorbed rapidly in the small intestine, result in an early peak plasma PHT concentration, and must be administered at least twice daily. Sustained-release products such as Dilantin Kapseals and Phenytek are the most commonly prescribed PHT products in the United States, deliver peak plasma concentration values 4 to 8 hours after administration, and may be administered once daily to adults. The liquid suspension is rapidly absorbed, has greater bioavailability than many capsule preparations, and also must be administered at least twice
daily.146
daily.146
Table 1 Hydantoins | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Prompt and sustained-release preparations differ sufficiently in pharmacokinetic properties that they cannot be interchanged. Most generic PHT products marketed in the United States are prompt preparations and cannot be substituted for sustained-release preparations. It should be noted that some preparations (e.g., Dilantin suspension or Dilantin Infatab chewable tablets) contain PHT acid, while others (e.g., Dilantin Kapseals and Phenytek) contain the sodium salt. Because there is approximately an 8% increase in drug content with the free acid form over that of the sodium salt, dosage adjustments may be necessary when switching from a product formulated with the acid to a product formulated with the sodium salt or vice versa.
Generic substitution of prompt preparations for sustained-release preparations is furthermore problematic because of three risk factors for inequivalence among generic forms of a drug: (a) poor water solubility, (b) nonlinear pharmacokinetics, and (c) narrow therapeutic range of plasma concentration.133,134 PHT possesses all three risk factors, so it is not surprising that comparisons of generic PHT products have yielded conflicting results, with more studies reporting inequivalence than equivalence.39,45,57,127,144,145,146,149,169,192 Two generic sustained-release PHT products were introduced in the United States and later withdrawn because of concerns that they may not be equivalent to brand-name Dilantin.15,64,169 Switching between preparations that are not equivalent may result in decreased drug plasma concentration (increased seizures) or increased drug plasma concentration (increased toxicity).20 Based on this information, an American Academy of Neurology position statement recommends that patients be managed with one form of PHT preparation supplied by one manufacturer.133,134
The oral suspension form of PHT is useful for patients who have difficulty swallowing capsules, especially children. The oral suspension also is useful in patients who do not absorb PHT capsules well.
Absorption by Intramuscular Route
Dissolving PHT in a small volume of liquid for parenteral administration requires a solution pH of 12. When this preparation is injected intramuscularly (i.e., into a medium with a pH of about 7.4), the water solubility of the drug decreases substantially, PHT crystals precipitate in the muscle,20 and the drug is absorbed very slowly.20,88
Peak plasma PHT concentrations occur approximately 24 hours after a single intramuscular injection and are considerably less than the peak concentration produced by the same dose given by intravenous infusion.88 PHT should not be given via the intramuscular route in emergency situations (e.g., status epilepticus) because of the slowness of absorption and the relatively low peak plasma concentrations produced by that route.
Use of the intramuscular route for administration of maintenance doses of PHT remains controversial. Eventually, almost all of an intramuscular injection of PHT is absorbed, and regimens for administration of maintenance doses of intramuscular PHT have been reported. However, peak plasma PHT concentrations after an intramuscular injection are variable.88 Furthermore, the plasma concentration may fall below therapeutic levels shortly after a switch to the intramuscular route from the oral or intravenous route and may ascend into the toxic range because of accumulation after switching back from the intramuscular to the oral route.88 In most situations, maintenance doses of PHT should not be given by the intramuscular route because of the slowness and variability of absorption and because of the danger of over- and undermedication when switching to and from other routes of administration.
Fosphenytoin is a water-soluble phosphate ester prodrug of PHT that eliminates many of the problems of sodium PHT for injection. Fosphenytoin is described below.
Plasma Protein Binding and Distribution
Plasma Protein Binding.
Distribution into Tissues and Other Body Fluids.
The concentration of PHT in brain parenchyma and cerebrospinal fluid (CSF) reaches peak values 15 to 60 minutes after intravenous injection in humans.189 However, the brain parenchyma concentration of PHT remains greater than the plasma concentration of free PHT once steady-state plasma concentrations are reached.
The concentrations of PHT in CSF, saliva, semen, and bile are essentially identical to the concentrations of free (non–protein-bound) PHT in plasma.189
Metabolism and Elimination
PHT is excreted in the urine and feces mainly as its metabolites, none of which has significant antiepileptic activity. Less than 5% is excreted in urine in an unmetabolized form.28,189 The major route of biotransformation is para-hydroxylation of a phenyl ring by the liver cytochrome P450 system (primarily isoforms CYP2C9 and CYP2C19) to form 5-(p-hydroxyphenyl)-5-phenylhydantoin (p-HPPH).6,28 Over 60% of a PHT dose is excreted in urine as p-HPPH, the majority of which is conjugated with glucuronic acid.28,189 Minor metabolites include a dihydrodiol derivative; meta-HPPH (mHPPH); diphenylhydantoic acid; 5,5-bis(4-hydroxyphenyl) hydantoin; 5-(3,4-dihydroxyphenyl)-5-phenylhydantoin; and 5-(3-methoxy-4-hydroxyphenyl)-5-phenylhydantoin.28,189
PHT is a prochiral compound. Introduction of a hydroxyl group in one of the phenyl rings leads to the creation of a chiral center and results in the formation of enantiomeric phenolic metabolites. The p-HPPH from human urine consists of a 10:1 mixture of levo- and dextrorotatory isomers.28,173 The amount of m-HPPH in human urine is too low to permit isolation and measurement of optical rotation. The majority of dihydrodiol in human urine is in the S configuration with varying S:R ratios.2,111,113,114,152 The S:R ratio is due to an approximately 40 times greater stereoselectivity of CYP2C9 for the S isomer and CYP2C19’s lack of stereoselectivity for either isomer.6
Occurrence of Nonlinear (Michaelis-Menten) Pharmacokinetics.
Nonlinear pharmacokinetic properties have major effects on PHT’s clinical usage. The rate of change of plasma concentration (C) of a drug metabolized by an enzyme system can be expressed by the Michaelis-Menten equation:
where t is time, Vmax is the maximum velocity of the enzyme system, and Km is the Michaelis constant of the enzyme system (plasma concentration at which half of the maximum velocity of the enzyme system is attained). The mean steady-state plasma concentration (Css) of the drug can be expressed as:
where R is the dosing rate. When C is similar to or greater than Km, dC/dt will vary in a nonlinear fashion with C; when R is equal to or greater than 0.1 × Vmax, Css will vary in a nonlinear fashion with R. These observations are the basis of nonlinear pharmacokinetics.
Based on 55 reported determinations, the mean apparent value for PHT’s Km in adults is 6.3 μg/mL and the range is 1.5 to 30.7 μg/mL. The mean apparent value for PHT’s Vmax in adults is 0.45 μg/mL/hr with a range of 0.14 to 1.36 μg/mL/hr based on 54 reported determinations.3,27,60,66,68,69,79,121,155 These values appear to be determined principally by arene oxidase enzyme system Km and Vmax values. The other minor pathways of PHT metabolism potentially could have modifying effects on the apparent values of Km and Vmax for PHT.108 However, attempts to demonstrate an effect of these other pathways on PHT pharmacokinetic parameters in humans have been negative.28,35
The apparent Km values computed for humans are based on total (protein-bound and non–protein-bound) PHT plasma concentration. Because only non–protein-bound PHT can be acted on by the metabolizing enzyme system and the non–protein-bound fraction for PHT is approximately 10% in humans,210 the Km of the enzyme responsible for para-hydroxylation of PHT actually should be approximately 0.6 μg/mL. This prediction has been verified in rat liver microsomes.193
Eadie et al.60 performed the first comprehensive comparison of PHT Km and Vmax values in children and adults. The Km values from 21 adults (mean 5.8 μg/mL) and 15 children (mean 5.3 μg/mL) were not significantly different. The Vmax values from 21 adults (mean 0.48 μg/mL/hr, assuming a PHT volume of distribution of 0.7 L/kg) were significantly (p <0.025) less than the Vmax values from 15 children (mean 0.74 μg/mL/hr assuming a PHT volume of distribution of 0.7 L/kg). Suzuki et al.67a,183 later confirmed these observations regarding Km and Vmax in children. These observations predict that the clearance [Vmax/(Km + C)] of PHT should be greater in children than in adults. This prediction is confirmed by the observations that the elimination half-life of PHT is shorter in children than in adults and that the average dosing rate of PHT (in milligrams per kilogram per day) required to achieve a given plasma concentration is greater in children than in adults.20,56,90
The Vmax for PHT increases significantly during pregnan-cy.54 This explains part of the observed decrease in PHT steady-state plasma concentration during pregnancy (Equation 2).
PHT will exhibit nonlinear pharmacokinetic properties in the majority of patients because the usual therapeutic plasma concentration values (10 to 20 μg/mL) exceed the usual Km (6.3 μg/mL), and the usual dosing rate (0.15 to 0.45 μg/mL/hr) is greater than 0.1 times the usual value of Vmax (0.45 μg/mL/hr). The consequences of nonlinear pharmacokinetics are discussed in the following sections.
Relationship between Plasma Concentration, Clearance, and Half-life.
According to the Michaelis-Menten equation, drug clearance is equal to Vmax/(Km + C). Drug elimination half-life is equal to 0.693 × volume of distribution/clearance. Thus, PHT clearance will vary inversely with plasma concentration, and PHT elimination half-life will vary directly with plasma concentration (Fig. 1).25,26,35 Browne et al.27,35 described and validated a method for calculating PHT elimination half-life at any given PHT plasma concentration if the patient’s Km and Vmax values for PHT are known. The results (plasma concentration and calculated elimination half-life) for a group of six adult men on PHT monotherapy were 1 μg/mL, 12.8 hours; 10 μg/mL, 25.8 hours; 20 μg/mL, 40.2 hours; and 40 μg/mL, 69.1 hours. Based upon these results, PHT’s commonly believed elimination half-life of 24 hours applies principally to plasma concentration values in the low therapeutic range (10 μg/mL) and the elimination half-life often is longer at higher plasma concentrations (also see Fig. 1). The range of elimination half-life values at a PHT plasma concentration of 40 μg/mL was 37.1 to 96.8 hours. Because of PHT’s long and variable elimination half-life values at toxic plasma concentration values, one cannot predict the time required for PHT plasma concentration to fall from a toxic value to a therapeutic value in a given individual. In such circumstances, one must withhold PHT and obtain daily plasma concentration determination values until the plasma concentration has fallen back into the therapeutic range.
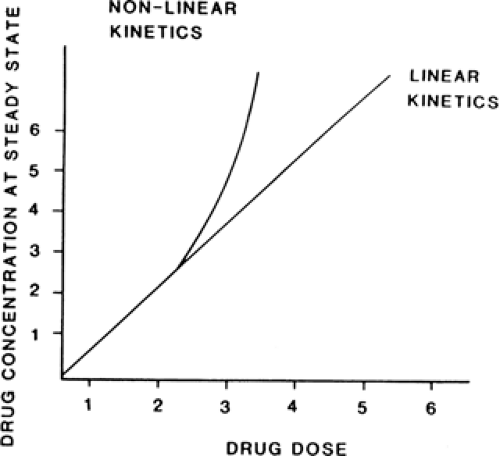 FIGURE 1. Relationship between dosing rate and steady-state plasma concentration for drugs with linear pharmacokinetics and for drugs with nonlinear Michaelis-Menten pharmacokinetics. |
Relationship between Steady-State Plasma Phenytoin Concentration and Dosage.
For drugs with Michaelis-Menten pharmacokinetics similar to PHT’s, steady-state plasma concentration increases to a greater extent than dosing rate
when dosing rate is increased, and plasma concentration decreases to a greater extent than dosing rate when dosing rate is decreased (Equation 2) (see Fig. 2).32,56,204 Thus, the steady-state plasma concentration of a drug with these Michaelis-Menten pharmacokinetics at one dosing rate does not directly predict the steady-state plasma concentration of the drug at another dosing rate.
when dosing rate is increased, and plasma concentration decreases to a greater extent than dosing rate when dosing rate is decreased (Equation 2) (see Fig. 2).32,56,204 Thus, the steady-state plasma concentration of a drug with these Michaelis-Menten pharmacokinetics at one dosing rate does not directly predict the steady-state plasma concentration of the drug at another dosing rate.
 FIGURE 2. Phenytoin clearance and elimination half-life values determined by stable-isotope tracer techniques at 30 different serum concentration values in 18 adult men on PHT monotherapy. (From Browne TR, LeDuc B. Phenytoin: chemistry and biotransformation. In: Levy RH, Mattson RH, Meldrum BS, eds. Antiepileptic Drugs. 4th ed. New York: Raven Press; 1995:283–300, with permission.) |
If the clinician attempts to increase or decrease PHT steady-state plasma concentration by simple linear extrapolation from a known plasma concentration versus dosing rate point, the result is often an unexpectedly high or low plasma concentration when the new steady-state value is attained (Fig. 2). Numerous mathematic and tabular methods have been published that claim to be able to predict the PHT dosing rate necessary to produce a given steady-state plasma concentration from a single steady-state plasma concentration versus dose point, and these methods have been critically reviewed elsewhere.23,32,131,138,139 A useful rule of thumb in titrating PHT dosage upward in adults is to increase dosing rate in increments of 100 mg/day at monthly (see below) intervals until a steady-state PHT plasma concentration of 5 to 10 μg/mL (a value approximately equal to Km) is attained; later increases should not exceed 50 mg/day at monthly intervals.
Nonlinear pharmacokinetics complicates the issue of generic equivalence of PHT products. The weighted mean value for absolute bioavailability of brand-name sustained-release PHT (Dilantin Kapseals, 100 mg, Parke-Davis) was 86% in three studies.20 Less than complete absorption of brand-name PHT is at least in part a consequence of the use of a sustained-release preparation (see above). Different generic preparations of PHT have the potential to differ in bioavailability from the brand-name preparation by 14% or more. Because of PHT’s nonlinear pharmacokinetics, a 14% difference in bioavailability is expected to result in a >14% increase or decrease in steady-state plasma concentration. For example, the bioavailability of a single dose of Mylan extended-release PHT given with a high-fat meal was found to be 13% lower than the bioavailability of Dilantin Kapseals taken under similar conditions.206 Pharmacokinetic simulations based on these data showed that substituting the Mylan product for Dilantin in patients with baseline plasma PHT concentrations within or above the optimal range would be expected to result in a median 37% decrease in PHT concentrations, whereas substituting Dilantin for Mylan would be expected to produce a median 102% increase in PHT concentrations. A national epidemic of PHT intoxication occurred in Australia when a more bioavailable formulation was substituted for an older formulation.154,199 Ludden et al.108 and Browne et al.36 have reviewed the effect of nonlinear pharmacokinetics on bioavailability studies in more detail.
Effect of Nonlinear Pharmacokinetics on Time to Reach Steady-State Plasma Concentrations.
According to the principles described above, as PHT plasma concentration rises, PHT clearance decreases and elimination half-life decreases (see Table 2 for typical values for these changes in a group of six patients started on PHT monotherapy). This results in a further rise in PHT plasma concentration and a further decrease in PHT clearance. This self-propagating cycle can require a long period of time to go to completion. The time (t) required to attain a given plasma concentration can be computed by the equation:
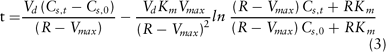
where Vd is volume of distribution, Cs,t is plasma concentration at time t, and Cs,0 is plasma concentration at time 0.204 Assuming average values for Km and Vmax, it is possible to compute an accumulation half-life (t1/2A) for PHT as follows:
where 1/2A has units of days and Css has units of micrograms per milliliter.94 Equations 3 and 4 predict, and empirical data confirm, the following: (a) time to reach steady-state plasma
concentration will vary nonlinearly with dosing rate; (b) time to reach steady-state plasma concentration will vary linearly with plasma concentration; and (c) the time required to attain new steady-state plasma concentration values after starting PHT therapy or increasing or decreasing PHT dosing rate may be as long as 28 days (Fig. 3).26,32,94,204 Therefore, in some individuals, particularly when the plasma concentration of the drug is in the high range, a PHT plasma concentration value measured <28 days after a change in PHT dosing rate may not be an accurate indication of the ultimate new steady-state
plasma concentration that will result from the change in dosing rate.
concentration will vary nonlinearly with dosing rate; (b) time to reach steady-state plasma concentration will vary linearly with plasma concentration; and (c) the time required to attain new steady-state plasma concentration values after starting PHT therapy or increasing or decreasing PHT dosing rate may be as long as 28 days (Fig. 3).26,32,94,204 Therefore, in some individuals, particularly when the plasma concentration of the drug is in the high range, a PHT plasma concentration value measured <28 days after a change in PHT dosing rate may not be an accurate indication of the ultimate new steady-state
plasma concentration that will result from the change in dosing rate.
Table 2 Phenytoin pharmacokinetic values for six patients at three times during monotherapy determined with 150-mg tracer doses of stable-isotope-labeled phenytoina | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
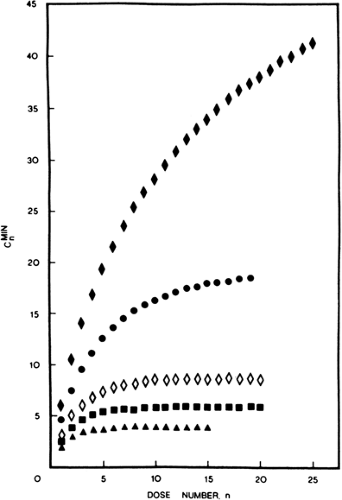 FIGURE 3. Plot of minimum serum concentration after the nth dose, Cmin, versus dose number n. Symbols and dosing rates (g/day) are: [black four-pointed star], 0.50; •, 0.40; ♦, 0.30; ▪, 0.25; and ▲, 0.20. (From Wagner JG. Time to reach steady state and prediction of steady state concentration for drugs obeying Michaelis Menten elimination kinetics. J Pharmacokinet Biopharmaceut. 1978;6:209–225, with permission.) |
Factors Affecting Plasma Phenytoin Concentration Versus Dose Relationship: Age.
Neonates eliminate PHT more slowly than adults or children (see below). Young children metabolize PHT more rapidly than older children and adults, requiring higher milligram-per-kilogram doses of PHT to achieve a given plasma concentration and sometimes not maintaining therapeutic plasma concentrations with once-daily administration.20,56 Plasma-unbound PHT concentrations for a given daily dose tend to be higher in the elderly than in young adults, probably because of a decreased metabolic rate, even though total plasma PHT concentrations may not differ to a major extent between young adults and elderly subjects.5,6a,62 Elderly individuals may also show a wide (two- to threefold) intraindividual variability in plasma PHT concentrations despite unchanged daily doses, possibly due to day-to-day variability in absorption efficiency.11
Factors Affecting Plasma Phenytoin Concentration Versus Dose Relationship: Pregnancy.
Decreasing total PHT plasma concentrations are usually noted throughout preg-nancy.102,147,186,187 However, the plasma concentration of free (unbound), pharmacologically active PHT falls much less than total PHT plasma concentration during pregnancy.186,187 Monitoring of free PHT plasma concentration may be advan-tageous during pregnancy.
Factors Affecting Plasma Phenytoin Concentration Versus Dose Relationship: Hepatic Insufficiency.
The oxidative metabolism of PHT and many other drugs is slowed in patients with hepatic disease.148 In addition, protein binding may be reduced by any of three occurrences: (a) hypoalbuminemia, (b) displacement by bilirubin or other substances, and (c) changes in the configuration of albumin. The overall result is usually an increase in the total plasma drug concentration, the free (unbound) drug concentration, or both.
Unfortunately, there is no formula for predicting the proper dose of an antiepileptic drug in a patient with hepatic dysfunction based on plasma albumin concentration or liver function tests. One must adjust drug dosage on the basis of clinical response and frequent determinations of the drug plasma concentration.
Factors Affecting Plasma Phenytoin Concentration Versus Dose Relationship: Renal Insufficiency.
Uremic patients receiving PHT have lower total plasma PHT concentrations, higher plasma p-HPPH concentrations, and shorter PHT elimination half-lives than nonuremic patients receiving the same dosage of PHT.72,98,123 In renal insufficiency, the hepatic biotransformation processes continue and may accelerate, but renal excretion of metabolites is slowed. The high plasma concentration of p-HPPH is presumably a result of impaired renal excretion of the metabolite. The short elimination half-life is presumably caused by increased accessibility of PHT to hepatic biotransformation enzymes as a result of decreased protein binding from low plasma albumin concentration, displacement of PHT from protein-binding sites by p-HPPH and endogenous metabolites, and structural alterations of plasma proteins.137 The low total plasma PHT concentration reflects primarily enhanced drug clearance as a result of increased accessibility of PHT to he-patic biotransformation enzymes. Because of the reduced PHT binding to plasma proteins and increased unbound fraction, total plasma PHT concentration in patients with uremia underestimates the concentration of free, pharmacologically active drug. Therefore, therapeutic and toxic effects occur in these patients at lower than usual total plasma PHT concentrations. In individuals with uremia, PHT therapy should be preferably monitored by measuring free (unbound) plasma PHT concentration.
Hemodialysis has little effect on the plasma concentration of PHT.105
Efficacy
Tonic–Clonic and Partial Seizures
The older literature on the effectiveness of PHT for tonic–clonic and complex partial seizures has been reviewed by Coatsworth.46 More recently, a series of trials has compared PHT with other older antiepileptic drugs (carbamazepine, phenobarbital, primidone, and valproic acid) as initial therapy in neonates, in children, and in adults with tonic–clonic and partial seizures.41,78,117,119,141,145,157,166,184,194,195,196,207 The results of these studies may be summarized as follows: (a) no drug was more effective than PHT in a randomized, prospective double-blind study; (b) PHT and carbamazepine were very similar in efficacy and toxicity; and (c) phenobarbital and primidone generally had more adverse effects than PHT or carbamazepine and, in some studies, were less effective when treating patients with partial seizures. There is also evidence-based data showing that PHT, oxcarbazepine, and lamotrigine have similar efficacy for the initial therapy of partial seizures.10,71,130,181 Although there have been no direct comparisons of PHT with topiramate and gabapentin, a recent trial suggested that topiramate may have inferior tolerability, and gabapentin inferior efficacy, compared with carbamazepine in patients with newly diagnosed partial seizures.114b Among newer generations antiepileptic drugs (AEDs), only oxcarbazepine and topiramate are approved by the Food and Drug Administration (FDA) for this indication.
In most of the above studies, tonic–clonic seizures were mostly secondarily generalized seizures in patients with focal epilepsy. However, PHT is also effective for primarily generalized tonic–clonic seizures.158,207 Although PHT is effective as prophylaxis against acute symptomatic seizures that may occur soon after traumatic brain injury, it does not decrease the risk of later developing remote symptomatic seizures or epilepsy.44
Status Epilepticus
PHT is an important therapy for status epilepticus manifesting with tonic–clonic or partial seizures. A randomized, double-blind trial of four treatments for status epilepticus found lorazepam to be more effective than PHT for seizure control at 1 hour, but it was equally effective to a combination of diazepam and PHT or phenobarbital alone.188 PHT was inferior to valproic acid for terminating convulsive status epilepticus in a randomized comparison trial of intravenous formulations with each medication given without benzodiazepines and with efficacy measured at the end of the infusion.129 However, valproic acid does not have FDA approval for this use.
Other Epileptic Disorders
Older studies, without adequate controls, yielded conflicting results in the value of PHT in treating alcohol withdrawal seizures.1 However, two modern and well-controlled studies strongly indicate that PHT has no value in treating or preventing alcohol withdrawal seizures.1,161
Other Indications
PHT controls trigeminal neuralgia in a smaller percentage of patients than carbamazepine, but the combined use of PHT and carbamazepine for trigeminal neuralgia is sometimes more effective than carbamazepine alone. PHT has been suggested as a therapy for a wide variety of organic and functional diseases of the nervous and cardiovascular systems, as well as for a variety of other conditions.13
Adverse Effects
Dose-related Central Nervous System Adverse Effects
The usual dose-dependent adverse effects of PHT include nystagmus, ataxia, and drowsiness.38,49 Nystagmus is usually horizontal but can include vertical movements at high drug plasma concentrations. The ataxia involves station and gait more than fine motor movements. There is an approximate correlation of signs of PHT intoxication with drug plasma concentration. In the majority of patients, nystagmus appears with drug plasma concentrations of about 20 μg/mL, ataxia with levels of about 30 μg/mL and drowsiness with levels of >40 μg/mL.90 However, there is considerable variation among patients in the plasma PHT concentration at which a symptom occurs, and not all patients experience all symptoms. At high plasma concentrations (usually >40 μg/mL), a reversible external ophthalmoplegia may occur.180 Patients also may experience an excited delirium rather than sedation with therapeutic and toxic PHT plasma concentrations.49
Perhaps the most important questions about PHT toxicity are concerned with what, if any, effects the drug has on cognitive function and behavior at typical therapeutic plasma concentrations. The cognitive effects of PHT have been studied extensively. Early studies were flawed by the fact that PHT slows motor speed, causing slowing of timed tests that is not related to slowed cognition. Recent studies indicate that (a) PHT has modest or little effect on cognition, (b) PHT and carbamazepine have similar and modest effects on cognition, and (c) barbiturates may have greater effects on cognition.38,47,55,117,124,176,203
A reversible syndrome of “PHT encephalopathy” has been described as a complication of chronic PHT therapy, usually with PHT plasma concentrations in the toxic range.49,163 The syndrome is characterized by mental changes, increased slowing and increased paroxysmal activity on electroencephalograms (EEG), increased seizure frequency, and a change in seizure pattern involving development of more tonic components. The mental changes may include drowsiness, progressive decline in higher intellectual function, depression, or euphoria. Focal neurologic signs such as hemiparesis and hemisensory defects may occur. Ataxia and nystagmus may be present or absent, and the CSF protein value may be elevated. This encephalopathy is more common in children, and pre-existing brain damage may be a risk factor for it.165 All these features generally disappear when PHT is discontinued.
Movement disorders (usually choreoathetosis and orofacial dyskinesia; rarely asterixis, ballismus, hyperkinesia, external ophthalmoplegia, periodic alternating nystagmus, or downbeat nystagmus) have been reported to be caused by PHT.49,160,180 These disorders usually occur with toxic PHT plasma concentrations but may occur with plasma concentrations in a typically therapeutic range for a smaller minority of patients.49
Murphy et al.131 reported 85 cases of PHT intoxication following unintentional overdose (PHT plasma concentration 30.3 to 95.0 μg/mL, median 46.5 μg/mL). The most frequent symptoms were nystagmus (95%), ataxia (80%), lethargy (22%), and seizures (19%). No patient had cardiac abnormalities attributable to PHT intoxication. Outcome was generally good, but three patients had serious complications (hip fracture, infections).
Dose-related Gastrointestinal Adverse Effects
PHT can cause nausea, vomiting, or constipation. Administration with or immediately after meals can reduce gastrointestinal discomfort.
Dose-related Cardiovascular Adverse Effects
Intravenous administration of PHT may cause hypotension, atrioventricular conduction block, and other dysrhythmias. These effects are related to dose and rate of administration, and conventional practice is to limit the rate of intravenous PHT infusion to 50 mg/min or less. Blood pressure and electrocardiographic (ECG) monitoring are recommended when PHT is administered intravenously, particularly at the higher dosages and infusion rates that are used for loading or for the treatment of status epilepticus. Some patients develop the cardiovascular side effects of the intravenous infusion even at the recommended maximum rate of 50 mg/min.
Chronic Adverse Effects
Effects on the Nervous System.
A loss of cerebellar Purkinje cells has been reported in association with PHT therapy.49,163 However, loss of Purkinje cells is a common finding in patients with epilepsy, whether or not they take antiepileptic drugs. Dam49 reviewed the human and animal data on the effects of PHT on Purkinje cells and concluded that PHT in therapeutic doses does not lead to changes in the density or substructure of Purkinje cells. Reynolds163 concluded that although much of the data on this topic are controversial, there are several convincing clinical reports of chronic cerebellar dysfunction following acute PHT intoxication. Since Reynolds’ review, additional reports of cerebellar atrophy following PHT intoxication have emerged38,116 as well as reports of PHT-related cerebellar degeneration in patients without seizures.135,159 Conversely, Murphy et al.131 found no evidence of permanent cerebellar dysfunction in 85 cases of severe PHT intoxication. Thus, permanent cerebellar dysfunction may be a rare complication of PHT intoxication and may be related to the duration of acute PHT toxicity or to pre-existing pathologic damage in the cerebellum from seizures, other drugs, or other causes.163,164
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


