Fig. 12.1
Image of the structure of PrPC showing three alpha helices, a single disulphide bond, up to two carbohydrate moieties and attachment to the cell surface via a glycosylinositolphosphate anchor. An N-terminal region containing octapeptide repeats appears to be unstructured and is not shown
12.2 Clinical Features of Prion Diseases
12.2.1 General Overview
Although there is considerable heterogeneity in the clinical picture of the prion diseases there are a number of core features. All are associated with cognitive decline at some stage of the illness which characteristically is rapid. This decline may remain focal for some time but ultimately becomes global. Memory, speech and executive functions are often involved early. Many patients are profoundly apraxic and some have complex articulation and language disturbances. The latter preferentially involves expression rather than comprehension in most cases and can be extremely prominent in some genetic types. Other features dependent on parietal function, such as getting lost in familiar surroundings and dressing apraxia, are frequent. Frightening visual hallucinations frequently occur but high order visual dysfunction is uncommon except in the Heidenhain variant of sporadic CJD. Executive dysfunction is common and often associated with behavioural change. The latter may require careful management and comprise irritability, aggression or withdrawal from normal social interchange.
Neurological symptoms and signs indicate involvement of multiple components in the nervous system, which is often the first clue to a prion disease vs. more common dementias. In the motor systems, ataxia, especially of gait, and dysarthria are early features in many types of CJD including variant CJD (vCJD), kuru, iatrogenic CJD (iCJD) and some types of sporadic CJD. Abnormal movements, especially myoclonus of cortical or subcortical type, is characteristic of most types of prion disease, especially sporadic CJD. Chorea is found in a proportion of sporadic and variant CJD, and exceptionally alien limb phenomena and epilepsia partialis continua have been reported. Stiffness with increased tone in the limbs and neck occurs in many types of CJD. This tone increase commonly has extrapyramidal features, especially in the upper limbs, in addition to spasticity. Fasciculation rarely occurs. Power in general is relatively preserved but hemiparesis or a stroke-like onset has been described.
Sensory loss is not often detected because the patients are frequently not capable of cooperating with the examination. Hyperaesthesiae is typical of vCJD and iCJD due to growth hormone and loss of thermal sensation occurs in some inherited prion disease. Sensory loss and autonomic failure is prominent in PrP-systemic amyloidosis associated with truncating mutations of PRNP.
Late in the course of all these diseases incontinence develops. Ultimately most patients enter a state of akinetic mutism. Death generally follows decreasing conscious level, pneumonia and respiratory failure or sepsis.
12.3 Sporadic CJD
Sporadic CJD (sCJD) is the most frequent type of spongiform encephalopathy in man. Although a rare disease with an incidence of 1–2 per million throughout the world, ascertainment in old age, when dementia is prevalent, remains an important unknown. The most frequent phenotype is of a rapidly progressive disease characterised by cognitive decline, visual disturbance, apraxia, ataxia and myoclonus. Typically the age of onset is between 55 and 70 years but many cases occur outside this range; cases in the 70–85 years range have probably been overlooked in the past but cases under the age of less than 45 years are rare. Only three cases of sporadic CJD younger than 30 years old have been identified in the UK since 1970. The disease affects males slightly less frequently than females. Death typically occurs in 5 months from onset in the majority, with only an atypical 10 % surviving up to 2 years, or exceptionally even longer, from onset.
Clinicians have long recognised different phenotypes of sCJD, that described by Jones and Nevin being the most frequent. Myoclonus is characteristic at some stage of the disease in virtually all cases but can be subtle. The Heidenhain variant is not infrequent and is characterised by visual disturbance culminating in cortical type blindness.
Other phenotypes include an ataxic variant, a thalamic variant and a panencephalitic type with extensive white matter change, these latter being mainly in the Japanese literature but may merely reflect long duration of disease where more grey matter is destroyed. It is uncertain if an amyotrophic type occurs and these cases, if they exist, have not been transmitted. Some case series describe groups with long pure cognitive and/or psychiatric phases early in the clinical course. Whether or not these different phenotypes represent distinct disease entities or extremes of a range of involvement of different neurological systems is not clear.
12.3.1 Diagnostic Criteria
The World Health Organisation have drawn up criteria for diagnosing various types of CJD and recently MRI criteria have been recommended to be added (Table 12.1). While these criteria are useful in epidemiological surveys ensuring uniformity of data, they may be restrictive in clinical trials where early diagnosis is essential.
Table 12.1
MRI-CJD consortium criteria for sporadic Creutzfeldt–Jakob disease
I- Clinical signs (with a symptom duration of less than 2 years) |
Dementia |
Cerebellar or visual |
Pyramidal or extrapyramidal |
Akinetic mutism |
II- Tests |
Periodic sharp wave complexes on the EEG |
14-3-3 protein detection in the CSF |
High signal abnormalities in caudate nucleus and putamen or at least two cortical regions (temporal, parietal or occipital) either in diffusion-weight imaging (DWI) or fluid attenuated inversion recovery (FLAIR) MRI |
Probable CJD |
Two out of I and at least one out of II |
Possible CJD |
Two out of I and duration less than 2 years |
12.4 Pathology
Macroscopically the cerebral hemispheres in sCJD are often of normal appearance although atrophy occurs in long standing cases. Cerebellar atrophy occurs in kuru, iCJD, vCJD and Gerstmann–Sträussler–Scheinker syndrome (GSS) and there is striking thalamic atrophy in familial fatal insomnia.
The characteristic microscopic features of prion diseases on haematoxylin and eosin staining are spongiform degeneration of the cerebral cortex, neuronal loss and gliosis associated with amyloid deposition in some cases (Fig. 12.2). Spongiform change begins in the dendrites, especially the presynaptic zones, and expands to form vacuoles in the neuropil. This is different to some animal prion diseases in which only the latter is usually apparent. Typically the vacuoles are 2–20 μm but can expand into much larger structures. The vacuoles have numerous membranous fragments in them on electron microscopy. In sporadic disease cortical layers four and five are most affected in the typical case with additional pathology in the basal ganglia, thalamus and cerebellum. Some of the inherited diseases have relatively little or no vacuole formation. There is substantial neuronal loss in most cases that increases with time.
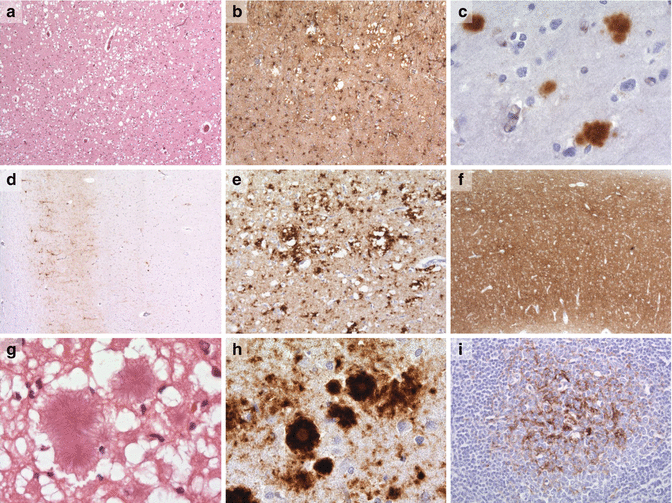
Fig. 12.2
Examples of prion pathology courtesy of Professor Sebastian Brandner, UCL Institute of Neurolgy. (a) spongiform change in sCJD (H&E), (b) gliosis and spongiform change in sCJD (GFAP), (c) kuru-like plaques in sCJD (ICSM35), (d) perineuronal PrP staining in sCJD (ICSM35), (e) perivacuolar PrP staining in sCJD (ICSM35), (f) synpatic PrP staining in sCJD (ICSM35), (g, h) florid plaques in vCJD (H&E, ICSM35), (i) PrP deposition in a tonsillar biopsy specimen in vCJD (ICSM35)
The distribution of pathology is dependent on the type of CJD. In vCJD and the insomnias thalamic damage is prominent while in Gerstmann Straussler Scheinker disease (P102L) the cerebellum is most involved. Neuronal loss and gliosis occur in a similar distribution. It should be emphasised that these changes are variable between patients even in those with the same phenotype.
Understanding the microscopic pathology has been greatly enhanced by the development of specific PrP immunostains, which not only demonstrate amyloid plaques better, but also show other abnormalities not apparent on routine stains. Amyloid plaques, comprising aggregations of PrPSc, occur in a variety of prion diseases. In sCJD they occur in about 10 % of patients; they have a dense core with a fibrillary halo, the so called kuru plaques (Fig. 12.2). Some inherited disorders such as GSS (P102L) have numerous plaques in the cerebellum which have a multicentric appearance while those patients with the 6-OPRI mutation have a pathognomonic linear arrangement in the cerebellum perpendicular to the surface. In contrast patients with familial insomnia have no amyloid plaques. vCJD patients have numerous large florid plaques with a characteristic pattern of fibrillary structure surrounded by spongiform change (Fig. 12.2).
Non-plaque deposition occurs in all cases of sCJD. The deposits can be granular synaptic, perineuronal decorating the neurones, or perivacuolar; the type of deposition depends on the subtype of sCJD and usually one type dominates (Fig. 12.2). In some cases there are so called mini-plaques which occur particularly in iCJD.
Tau inclusions are common in all types of CJD and are particularly prominent in vCJD in spite of the young age of the patients. Tau co-localises with prion amyloid. The morphology of Tau deposits is different to that seen in Alzheimer’s disease. In the latter the deposits are thread like whereas in CJD they form minute rods.
The pathology in the spinal cord and nerve roots is less well documented. Few autopsy studies have been done in sCJD but in iCJD with P102L mutation there is loss of fibres in the corticospinal, spinocerebellar and gracile tracts. Patients carrying the E200K mutation sometimes have a mixed axonal and demyelinating sensori-motor neuropathy.
While the pathology in sCJD, the insomnias, inherited disease and some of the iatrogenic disease (growth hormone and dural implantation) the abnormalities are confined to the CNS including the spinal cord, this is not the case with vCJD, or PrP systemic amyloidosis. These diseases have systemic pathology, notably in vCJD in the lymphoreticular system, including the spleen, and in this respect are similar to cervid chronic wasting disease (CWD), ovine and caprine scrapie, and to a lesser extent BSE. Prion staining is positive early in the course of the disease in animals and precedes the encephalopathy, a situation that almost certainly applies to man. The deposition in the lymphatic tissue is the basis of tonsil biopsy for diagnosis in vCJD where all definite cases have been positive (Fig. 12.2).
PrP systemic amyloidoses are a group inherited prion diseases, of which Y163X is the best documented, and are characterised by vascular and parenchymal PrP amyloid with a broad distribution in the CNS and systemic organs. These patients present with diarrhoea, autonomic failure, sensory neuropathy and urinary incontinence.
12.4.1 Molecular Classification
Since the advent of prion protein gene (PRNP) analysis and prion protein analysis by Western blotting, a molecular classification of sporadic CJD has emerged. Although the subdivision of sCJD in this way is of interest from a research perspective, to date, it has little clinical relevance. Nevertheless some new facts have emerged. Firstly, a polymorphic genetic variant at PRNP codon 129 between methionine and valine, is one factor that is particularly important in determining the clinical phenotype and type of PrPSc observed by Western blot. Second, two classifications have been developed in which the genotype at codon 129 is combined with prion protein electrophoretic pattern on Western blot after partial digestion with protease viz that of Parchi and colleagues, and that of the London (MRC Prion Unit). The patterns of PrP staining after Western blot (Fig. 12.3) are different because of differential protease cleavage and differing predominance of three glycosylation states. The situation is further complicated by the sensitivity of Western blot protocols to changes in the experimental conditions and the frequent coexistence of types when multiple brain areas are examined.
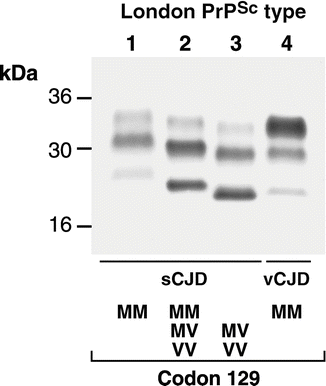
Fig. 12.3
Western blot (left) of four patient brain samples prepared by homogenization in phosphate buffered saline, partial protease digestion using proteinase K, and immunoblotting. Three PrP immunoreactive bands are seen related to three glycosylation states (un-, mono-, and diglycosylated). Types 1–3, distinguished by molecular weight, are seen in sCJD with restriction to certain codon 129 genotypes (below). Type 4, distinguished from sporadic types by the predominance of the diglycosylated (top) band, is exclusively seen in vCJD
As a starting point in the following discussion the London classification will be used and this is compared to the Parchi typing. In general, homozygosity at codon 129 causes a more aggressive disease. Type 1 patients who are 129MM tend to have the classical CJD phenotype with a mean age of 55–60 years and survival of weeks. Type 2 patients who are 129MM differ in that the course of the disease is usually longer (a few months) and they are younger with a mean age of onset of about 50 years, although ascertainment may be an influence here. In the Parchi classification there is no distinction between London types 1 and 2, thus giving a wider range of age and duration in their type MM1.
The most frequent type of sCJD in the London series is type 2 MM comprising some 45 % of all cases while about 16 % were type 1MM. The number of homozygous VV cases is smaller in all series making accurate description of the clinical picture less reliable. Certain rare forms of sCJD appear to be most distinct from the others. Heterozygosity at codon 129 causes a less aggressive disease in general, with longer survival, and the patients can initially mimic other neurodegenerative disease such as the frontotemporal dementias or have a marked apraxia at onset, and little or no myoclonus.
One final, but very rare, form of sCJD is sporadic fatal insomnia, a condition that is associated with autonomic features. This condition is characterised by thalamic involvement. There is a wide age range with a mean of about 50 years and duration of 16 months. The cases are classified as Parchi MM2 (thalamic) type.
A new type of sporadic prion disease was described in 2008 and 2010 termed variably proteinase sensitive prionopathy. These patients have clinical courses of 6 months – 4 years, and a predominance of behavioural and cognitive signs, compared with motor or cerebellar features. The condition also differs from sporadic CJD in its histopathological and immunoblotting features, with a ladder like electrophoretic profile on Western blot.
Development of a molecular classification for human prion disease may have implications for epidemiological research into the causes of sporadic CJD, whose aetiology remains obscure. Spontaneous conversion of PrPC to PrPSc as a rare stochastic event, or somatic mutation of PRNP, resulting in expression of a pathogenic PrP mutant are plausible explanations for sporadic CJD. However other causes for at least some cases including environmental exposure to human prions, or exposure to animal prions may also be important. In this regard, the number of prion strains causing sheep scrapie has yet to be established and epidemiological data cannot exclude this as a cause of a minority of cases. As future research begins to provide a more precise understanding of the origins of human prion disease, this will facilitate re-analysis of epidemiological data, and is likely to reveal important risk factors that might have been obscured by analysing sporadic CJD as a single entity.
12.5 Inherited Prion Disease (IPD)
There are at least 35 different pathological mutations causing inherited prion disease (IPD, Fig. 12.4). There are broadly three types of mutation viz: alteration of the normal number of a nonapeptide followed by four octapeptide repeats between codons 51 and 91 of PRNP, or point mutations in the C-terminal portion of the protein, and premature truncation of the protein by stop codon mutations.
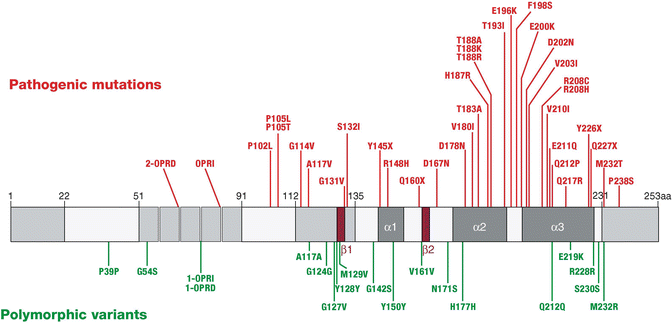
Fig. 12.4
Pathogenic mutations (red above) and polymorphic changes (green below) in the prion protein gene are shown on this schematic. The central grey bar also illustrates the secondary structural features of the prion protein
Phenotypes can be highly variable even in patients with the same mutation and within a family. Some of these cases have been transmitted to other species but many have not, particularly those reported from a single family. Failure of transmission in experimental situations does not necessarily mean that transmission will not occur given the appropriate route of inoculation and genetic background of the recipient. A summary of the mutations so far described is shown in Table 12.2.
Table 12.2
In this table the mean age (years) of onset, range of ages and clinical features of IPD mutations is shown
PRNP mutation | Median onset | Range | Clinical features | Modification by codon 129 genotype | Transmission of disease to laboratory animals |
|---|---|---|---|---|---|
P102L | 50 | 25–70 | GSS, CJD, psychiatric presentations, heterogenous | Possible | Yes |
P105L | 45 | 38–50 | GSS, spastic paraparesis | Not known | Not known |
G114V | 22 | 18–27 in one family | GSS, Neuropsychiatric and extrapyramidal signs prominent | Not known | Not known |
A117V | 40 | 20–64 | GSS with early cognitive, neuropsychiatric, extrapyramidal features | Yes | No |
G131V | 42 | GSS | Not known | Not known | |
Y145X | 38 | Alzheimer-like dementia | Not known | No | |
R148H | 72 | 63–82 | CJD | Not known | Not known |
Q160X | 40 | 32–48 | Unspecified dementia | Not known | Not known |
D178N | 50 | 20–72 | FFI, CJD | Possible | Yes |
V180I | 74 | 58–81 | CJD | Not known | Not known |
T183A | 45 | 37–49 | Prominent behavioral abnormalities, one patient with dementia and no additional signs | Not known | Not known |
Y163X | 30 | 30–50 | Early diarrhea and autonomic neuropathy Late development of CNS symptoms | Not known | No |
H187R | 32 | 20–53 | Early onset with personality disorder in one family
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





