PRION THEORY AND BIOLOGY
Cellular Prion Protein
The native cellular prion protein (PrPc) is located throughout the human body, predominantly in the brain and gut. PrPc is a gly- cosyl phosphatidyl inositol-linked glycoprotein that is anchored on lipid rafts. The tertiary structure of PrPc is primarily composed of a-helixes with few β-sheets. The prion protein gene (PRNP ) was identified in 1986 and its identification has led to the development of PRNP knock-out mice, which do not appear to behave differently from wild-type mice. However, PrPc null mice exhibit impaired synaptic inhibition and it has been proposed that PrPc has stress- protective functions. Divalent cations including copper, zinc and manganese have also been shown to bind to PrPc, though the significance of this is unclear. There has been emerging evidence that PrPc may be associated with Alzheimer’s disease and psychiatric illnesses as part of the stress diathesis.
Pathological Prion Protein
In contrast to the native prion protein, the pathological prion protein’s (PrPres) structure is primarily composed of β-sheets that aggregate and form amyloid plaques. The high β-sheet content of PrPres leads to its partial resistance to proteinase K, resulting in proteinase- resistant protein fragments (i.e. PrPres) called prions, which stands for ‘proteinaceous infectious particle.’
There are three aetiologies of PrPres : (i) sporadic, (ii) genetic and (iii) acquired. Although the exact mechanism for the formation of sporadic PrPres is unknown, it is widely believed to be the result of a posttranslational mutation. This has recently been demonstrated using in vitro protein misfolding cyclic amplification (PMCA) techniques1. PrPres can also be generated de novo by mutations within the prion protein gene (PRNP ). Lastly, PrPres can be acquired from an outside source and introduced into the body, precipitating the formation of additional PrPres.
Prion Theory (The Protein-Only Hypothesis)
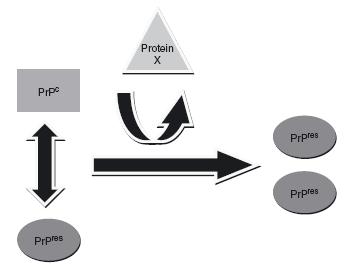
Prions are transmissible agents that lack the nucleic acids that bacteria and viruses use to infect other organisms. In lieu of nucleic acids, the transmissibility of prions is accomplished via an autocatalytic mechanism, a theory coined the ‘protein-only hypothesis’ (Figure 59.1). This process was first postulated by the mathematician J. S. Griffith in 19672 and was later described by Stanley Prusiner, who was subsequently granted a Nobel Prize for his work3. PrPres interacts with PrPc, using itself as a template to change the conformation of PrPc into another PrPres protein. Mouse studies have found that the presence of PrPc is a requirement for the conversion process to occur. As evidence of this fact, PrPc-null mice do not develop prion disease when they are inoculated with PrPres, as the conversion process does not occur without the PrPc substrate.
Figure 59.1 The prion conversion process. PrPres replicates via an autocatalytic mechanism. PrPres uses PrPc as a substrate for the production of further PrPres proteins. An uncharacterized protein (protein X) is postulated to facilitate the transformation process
Kuru
Human prion diseases, or transmissible spongiform encephalopathies (TSEs), were initially described in detail by Nobel Prize laureate Carlton Gajdusek through his research on kuru4. Believed to be a ‘slow virus’, kuru was an endemic illness in the Fore tribe of Papua New Guinea. Kuru was characterized by rapidly progressive ataxia, oculomotor impairment, myoclonus and dementia with death typically occurring within two years of the illness onset. Gajdusek later found that kuru was transmitted among the tribe via its cannibalistic rituals of consuming their deceased, particularly their brains. Tribal members who consumed brain matter of relatives with kuru were exposed to the illness and incubated it for several years, sometimes decades, until they eventually developed the illness. It is generally accepted that the kuru epidemic has since subsided and no longer exists.
Creutzfeldt-Jakob Disease
The most common human prion disease is Creutzfeldt-Jakob disease (CJD), which was first described by two neuropsychiatrists, Hans Gerhard Creutzfeldt5 and Alfons Maria Jakob6. Both Creutzfeldt and Jakob described patients with a rapid neurodegenerative illness who had a spongiform appearance of their brain at postmortem analysis. CJD is recognized at autopsy by the neuropathological triad of astrogliosis, spongiform changes and neuronal death (Figure 59.2a). Immunostaining with monoclonal 3F4 antibodies also shows prion deposition within the brain (Figure 59.2b). Abnormal prion proteins can also be detected by pre-treating brain homogenate with proteinase K and performing Western blot analyses that typically detect proteinase-resistant protein fragments (PrP27-30).
Figure 59.2 (a) Haematoxylin and eosin (H&E) stain of the temporal cortex and (b) monoclonal 3F4 antibody immunostaining of the hippocampus in a patient with Creutzfeldt-Jakob disease. The neuropathology is characterized by a spongiform appearance, astrogliosis and neuronal cell death. Immunostaining (b) reveals abnormal prion protein deposits (dark brown). A full colour version of this figure appears in the colour plate section

CJD is a rare cause of dementia. The worldwide incidence of CJD is one per million people per year and is slightly higher in females than in males, although the age-adjusted incidence is slightly higher in males. CJD is rare in blacks for unclear reasons, but ascertainment problems may be one reason for the low incidence. Researchers have discovered a genetic propensity towards the development of CJD in relation to the codon 129 polymorphism of the PRNP gene. Individuals who are homozygous (Met-Met or Val-Val) at codon 129, especially Met-Met, are more likely to develop CJD. The PRNP codon 129 polymorphism is also associated with differences in clinical presentations and survival times.
CJD can be divided by aetiological subtypes. There are three main aetiologies of CJD: (i) sporadic CJD (sCJD), (ii) genetic CJD (gCJD), and (iii) acquired forms of CJD. The latter group includes the iatro- genic transmission of CJD (iCJD) and human transmission of bovine spongiform encephalopathy (BSE) called variant CJD (vCJD). In addition to different aetiologies, these subclasses of CJD also demonstrate different demographic, clinical, diagnostic, neuropathological and molecular characteristics. Table 59.1 lists the characteristics of the CJD subtypes7–8.
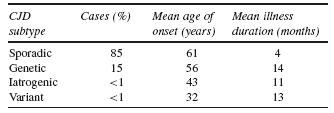
Figure 59.3 Age of onset of Creutzfeldt-Jakob disease. The age of onset follows a bell-shaped curve with a mean age of onset in sporadic CJD (sCJD) of 61 years, 56 years in genetic CJD (gCJD), and 32 years in variant CJD (vCJD). Adapted from Appleby et al.8
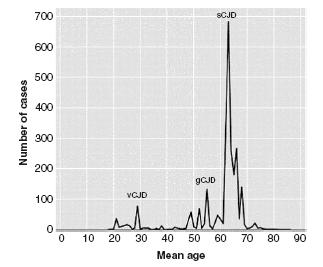
Table 59.2 Presenting symptoms of sporadic Creutzfeldt-Jakob disease
Source: Adapted from Appleby et al.8
| Presenting symptom | Cases (%) |
| Cognitive impairment | 72 |
| Ataxia | 44 |
| Visual/oculomotor impairment | 35 |
| Mood disorder | 28 |
| Vertigo | 26 |
| Behaviour/personality change | 23 |
| Psychosis/agitation | 20 |
| Sleep disorder | 18 |
Sporadic Creutzfeldt-Jakob disease
The exact aetiology of sCJD is unknown, but is largely believed to be a consequence of a posttranslational mutation of PrPc. Comprising 85% of all human prion diseases, sCJD is the most common human prion disease. The age of onset follows a bell-shaped curve with a mean age of onset of 61 years (Figure 59.3
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
 Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


