Figure 19.1. EEG–EMG recording with surface electrodes in a 3-year-old boy with MAE (Doose syndrome). Three brief and symmetric myoclonic jerks each time-locked to single generalized single spike–wave discharges are recorded from both deltoid muscles.
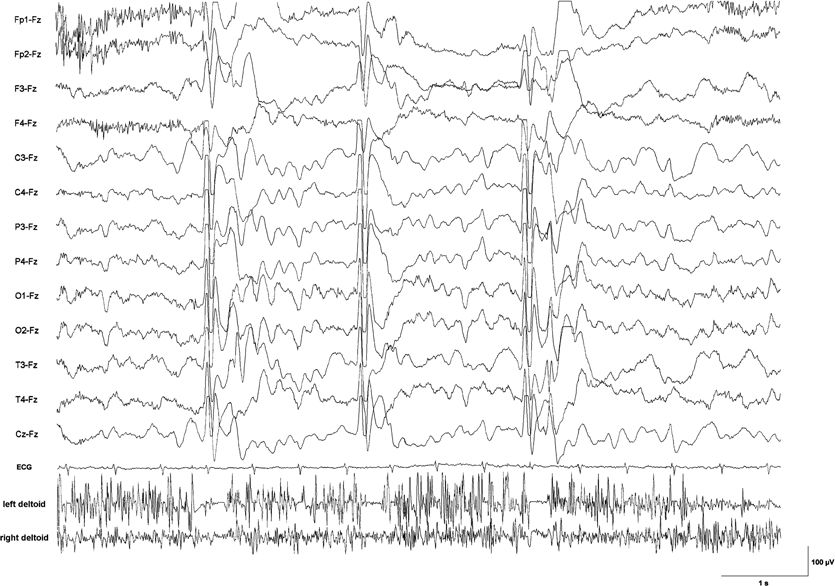
Figure 19.2. EEG–EMG recording with surface electrodes in a 6-year-old boy with epileptic negative myoclonus due to lesion-related epilepsy. Silent periods in the EMG from the deltoid muscles lasting between 100 and 150 ms are time-locked to a generalized sharp slow-wave complex. Clinically, each brief EMG pause was associated with nodding of the head and sinking of the arms during sustained muscle contraction.
ETIOLOGY
Myoclonic epilepsies are predominantly genetic in origin. In terms of classification, they may be grouped as genetic epilepsies (e.g., benign myoclonic epilepsy in infancy [BMEI]), epileptic encephalopathies (e.g., Dravet syndrome), or progressive myoclonic epilepsies (e.g., Unverricht–Lundborg disease).
BMEI usually presents in normally developed children, and in the majority of cases, antiepileptic treatment may be weaned after some years of active epilepsy. Behavioral and cognitive prognosis is good in the majority of cases. Familial cases are extremely rare, with the exception of one case identified in the context of a generalized epilepsy with febrile seizures plus (GEFS+) family (2) and a family with three affected children (3). However, a positive family history for febrile seizures or idiopathic epilepsies was repeatedly reported (4). Altogether, classification as a genetic generalized epilepsy syndrome seems unequivocal. The etiology is most likely oligogenic or complex (several genes and possibly environmental factors involved).
Myoclonic astatic epilepsy (MAE) or Doose syndrome, initially described by Doose et al. (5), was reported to be an idiopathic generalized epilepsy syndrome. The separation of this syndrome from a group of epilepsies that were formerly classified as symptomatic (many of them as Lennox–Gastaut syndrome) was only reluctantly accepted. This is mirrored by the fact that MAE was classified among the “cryptogenic or symptomatic generalized epilepsies and syndromes” for many years. Now it is recognized that the initial description of Doose et al. included cases that, from today’s point of view, might also be diagnosed as benign myoclonic and severe myoclonic epilepsies, but the current classification scheme now accepts Doose syndrome as a genetic epilepsy syndrome (6). As with BMEI, familial cases are exceptional, but retrospective studies demonstrated positive family histories for febrile seizures and idiopathic epilepsy syndromes at a clearly elevated rate (7). Although in single cases genetic defects in the genes (mostly SCN1A) known to be involved in GEFS+ families were reported, the majority of patients do not carry an identifiable defect in these genes (8).
Severe myoclonic epilepsy of infancy (SMEI) or Dravet syndrome from one point of view may be classified as an idiopathic genetic disorder, since children are healthy and normally developed until onset of the epilepsy, and there is a clear genetic cause (usually a SCN1A defect) in the majority of cases. The syndrome may even be observed in GEFS+ families (9). However, Dravet syndrome is a devastating disorder with an extremely severe course in most patients; hence, it was declared an epileptic encephalopathy (6).
Progressive myoclonus epilepsies (PME) are defined as neurometabolic or neurodegenerative diseases with myoclonias and myoclonic seizures as dominating clinical features. Frequently, myoclonic seizures represent the initial clinical symptom. These disorders are in general progressive; however, course varies from moderately mild (e.g., Unverricht–Lundborg) to rapidly progressive and fatal (e.g., neuronal ceroid lipofuscinoses [NCL] type 2). PME follow a monogenic, mostly autosomal recessive mode of inheritance (10). Some diseases such as Rett-like syndrome (CDKL-5 defects) are usually not regarded as “classic” PME but may at certain stages and in individual patients show the typical symptomatology and course of a PME. These disorders, which frequently manifest during infancy or early childhood, may be summarized as progressive encephalopathies with myoclonic seizures (Table 19.1).
Table 19.1 Progressive Myoclonic Epilepsies and Progressive Encephalopathies with Myoclonic Seizures

BENIGN MYOCLONIC EPILEPSY IN INFANCY
Definition and Epidemiology
BMEI is a rare epilepsy syndrome. Its classic description was done by Dravet and Bureau (11). Altogether about 110 cases have been reported, and it is estimated that BMEI accounts for <1% of childhood epilepsies (12). BMEI is mainly recognized in Europe (France, Italy), and it may be suspected that in cases with rare myoclonic seizures or seizures that respond quickly to therapy, correct classification may not be applied, thereby underestimating its real prevalence. Some cases may overlap with MAE (Doose syndrome). In one patient who was initially diagnosed with BMEI, later a SLC2A1 (glucose transporter-1) defect was established (4).
Symptomatology
Onset is mostly between 4 months and 3 years but may extend up to the 5 years. Febrile seizures antecedent to myoclonic seizures are reported in about 30% of cases. In the beginning, myoclonic seizures are often rare and brief, involving predominantly the upper limbs. But intensity and frequency of the seizures increase often early during course. In the largest series reported, polygraphic video–EEG recordings revealed that myoclonic seizures consisted of axial jerks with head drop, elevation and extension of both arms, flexion of the legs, and upward gaze. Myoclonic seizures may vary in intensity ranging from simple head nods to severe falls, when generalized. Seizures may occur repetitively but usually only in short trains lasting 1 to 3 seconds. Usually they are symmetrical, but rarely may be unilateral. In about a third of patients, they are triggered by stimuli-like noise, startle, or photic stimulation. Drowsiness is also known to provoke myoclonic seizures (4).
EEG
Background activity is normal. Myoclonic jerks are associated with generalized spike–waves and polyspike–waves. Ictal spikes last for 1 to 3 seconds. Most, if not all, myoclonic seizures are associated with discharges on the surface EEG. EEG discharges are generalized with a frontocentral accentuation. After the myoclonia, there may be a short atonia that may result in a drop, that is, a myoclonic astatic seizure. Focal abnormalities, usually spike–waves in sleep recordings, were reported in some patients (4). Its significance is unknown.
Treatment and Prognosis
Valproic acid is the drug of first choice and is usually the only drug needed to control the seizures in the majority of cases. Some authors recommend high plasma levels at the start of treatment (up to 100 mg/L) (13). Ethosuximide, benzodiazepines, and phenobarbital were also effective in the few cases reported who did not receive valproic acid. Altogether there seems to be no specific difference in treatment compared to MAE. Untreated cases continue with pure myoclonic seizures even if the epilepsy lasts for years. Developmental delay and behavioral disturbances are reported in a substantial number of patients. Precise numbers vary substantially between different series, ranging from 0% to 86% (14). A rate of 20% to 30% seems acceptable. This renders the term “benign” questionable and makes it difficult to differentiate cases with frequent generalized myoclonic seizures and evolving developmental delay from patients with MAE. Bureau and Dravet are convinced that mental prognosis depends on early diagnosis and successful treatment (4). However, there are no controlled studies with antiepileptic drugs (AED) on record, and there are no good data on required treatment duration. By definition, myoclonic seizures will disappear eventually in all cases. Most reported children older than 6 years were already weaned off medication without seizure relapse. But in some patients, generalized tonic–clonic seizures (GTCS) occurred when valproate treatment was stopped. While a subgroup with overt reflex seizures appears to be very easily controlled by AED, cases with marked photosensitivity may be more difficult to control (4).
MYOCLONIC ASTATIC EPILEPSY/DOOSE SYNDROME
DEFINITION AND EPIDEMIOLOGY
The prominent genetic etiology together with the characteristic seizure symptomatology dominated by myoclonic and myoclonic astatic seizures led Doose et al. (5) to delineate MAE as an idiopathic (today: genetic) generalized epilepsy syndrome of its own right. MAE, as a rule, occurs in children with an uneventful history. The epilepsy starts in 94% during the first 5 years of age and accounts for 1% to 2% of all childhood epilepsies (15). Doose et al. reported that in about 20%, the seizures have their onset during the first year of life (7,16). Today, many authors feel that onset before the second year of life is exceptional. The age of peak presentation is 3 years (17). Like most other myoclonic epilepsies of early childhood, it affects more boys than girls. The sex ratio is about 2.7/1 (16). If the inclusion criteria include all children older than 1 year, this ratio might even reach 3/1 (17).
Symptomatology
In about 60% of cases, the epilepsy starts with febrile or afebrile GTCS. Alternating grand mal (i.e., hemi–grand mal, unilateral seizures) is a possible presentation. Some days or short weeks later, myoclonic and/or myoclonic–astatic seizures set in abundance, frequently in combination with brief absences. At first, this occurs predominantly after awakening. Tonic axial seizures may manifest during long-term course, frequently occurring during sleep. Myoclonic seizures consist of symmetric, mostly generalized jerks, accentuated in the arms and the shoulders, and are frequently associated with a simultaneous flexion of the head. The intensity of these seizures is variable and ranges from violent myoclonic jerks with sudden falls to abortive forms merely presenting as short irregular twitches of the face. Myoclonic–astatic seizures are characterized by a loss of muscle tone preceded by a (short) myoclonia. In polygraphic recordings, the loss of muscle tone corresponds to a silent period in the EMG that is paralleled by the slow wave in the EEG following the spikes or polyspikes of the myoclonus. Myoclonic seizures with and without discernable myotonia frequently occur together. The initial myoclonia and the subsequent myotonia equally contribute to the characteristic myoclonic astatic seizure (18–20). Absences are seen in more than half of the children with MAE. Myoclonic and astatic seizures, when they come in series, are frequently accompanied by absences often combined with myoclonic jerks. A unique type of nonconvulsive status epilepticus (“status of minor seizures”) is a rather specific finding observed in 36% (5,7,18) to 95% (17) of MAE patients. The characteristic clinical picture is a somnolent, stuporous child with subtle myoclonic seizures, frequently involving the face and the extremities. The child is unresponsive, drools, has a slurred speech, or is even aphasic. This status may continue for days if not interrupted by adequate means.
EEG
Background activity is of special interest in MAE. In cases starting with GTCS, the EEG may stay entirely normal for weeks. However, almost in all instances, a rhythmic, parietally accentuated 4- to 7-Hz activity develops early in the course. This rhythmic slowing of background activity was frequently questioned and falsely attributed to drowsiness. In patients with MAE (and other idiopathic generalized epilepsies of early childhood with myoclonic seizures), it represents a constant trait that is not related to the state of vigilance. This has been documented by EEG recordings of children who were kept attentive by displaying cartoons and so on (20). During the early stages, spikes, irregular spikes, and polyspikes may well be absent and appear only after some delay starting during sleep. If myoclonic seizures dominate the course at a given time, the EEG shows short paroxysms of irregular spikes and polyspikes. In children with astatic and myoclonic astatic seizures, 2- to 3-Hz spikes and waves appear. As the epilepsy progresses, typical absence patterns may appear. During a status of myoclonic–astatic seizures, the EEG displays continuous spike–waves with interposed slow waves. Especially in younger children, an irregular polymorphous hypersynchronous activity, sometimes resembling hypsarrhythmia may be recorded. During nocturnal tonic seizures, typical 10- to 15-Hz spike series can be observed. In distinction to the tonic seizures observed in Lennox–Gastaut syndrome, the EEG onset is generalized in MAE.
Therapy and Prognosis
Revised treatment standards over the last years have significantly improved outcome and prognosis (21). Valproic acid is still the drug of first choice. If efforts fail to achieve complete remission, the decision which drug to use next depends on the predominating seizure type. If absences prevail, ethosuximide should be commenced as the next step. If GTCS represent the leading semiology, bromide is frequently the most effective drug, even superior to phenobarbital or primidone (22). Lamotrigine may be effective but is also known to provoke myoclonic seizures in generalized myoclonic epilepsies. It therefore may represent a valuable option but has to be used with caution (23). Basing on the broad mechanism of action, topiramate is an additional possibility. However, no data on its effectiveness in MAE are yet available. Carbamazepine, phenytoin, and vigabatrin should be avoided, for they frequently provoke seizure exacerbation (17,24,25). In cases with refractory nonconvulsive status epilepticus adrenocorticotropic hormone (ACTH) or high-dose steroid pulse therapy may be alternatives to be considered (24). Zonisamide is effective in myoclonic epilepsies of different etiology (26). Levetiracetam may be used successfully in myoclonic epilepsies; however, it may also aggravate seizures (27). Ketogenic diet is a further possibility that was reported as effective (28).
Already from the early descriptions of the disorder, it becomes clear that outcome is highly variable. The spectrum ranges from complete remission (frequently obtained within the first 3 years) and totally normal intellectual development to therapy resistant epilepsy with severe cognitive disability (5,21,24,29). Over the years, however, therapeutic possibilities constantly improved, and the danger of seizure and epilepsy aggravation by carbamazepine and phenytoin was more and more recognized. In a series of 81 patients with MAE, 68% became eventually seizure free. In this retrospective Japanese series, ACTH, ethosuximide, and ketogenic diet proved especially effective (21). Repetitive nonconvulsive status epilepticus (“status of minor seizures”) and nocturnal tonic seizures were frequently associated with an unfavorable prognosis (16,21). This is, however, not unequivocal (17).
SEVERE MYOCLONIC EPILEPSY OF INFANCY/DRAVET SYNDROME
Definition and Epidemiology
This electroclinical syndrome was delineated by Dravet (30) in 1978 and soon was labeled SMEI. Later, it was recognized that in a substantial number of cases, myoclonic seizures and single other features may be lacking, and still the epilepsy will take the same course. This led to the proposal to rename the epilepsy to Dravet syndrome, which is now recognized as an epileptic encephalopathy (6). In order to include cases that seemed to belong to the same entity but lacked single features of classic SMEI, recently the term “borderline SMEI” (SMEB) was introduced. Different variants of what is now called SMEB were earlier recognized independently by different authors, such as “intractable childhood epilepsy with GCTS,” “severe idiopathic epilepsy of infancy with generalized tonic–clonic convulsions,” “severe polymorphic epilepsy of infants,” and even a few others (8).
The rate of cases with Dravet syndrome seems to augment constantly as the syndrome may be diagnosed increasingly frequently by SCN1A (α subunit of the neuronal type I sodium channel) gene analysis (31,32). This unique opportunity has sharpened the diagnostic view of the medical community. After one has succeeded in diagnosing a few cases, it frequently becomes an experience of pattern recognition. Therefore, the formerly calculated incidence of 1/40,000 may be an underestimate (33). It is of note that many epileptic vaccine encephalopathies in which an immunization-provoked fever triggered the epilepsy were retrospectively identified as having a SCN1A mutation (34).
Symptomatology
The disease most frequently starts with febrile seizures within the first year of life, in an up to then healthy child. These seizures may initially be indiscernible from regular febrile seizures but frequently become prolonged. Fever, infections without fever, vaccinations, hot baths, or even only hot weather may trigger recurrent seizures. These may occur generalized or unilateral affecting different sides of the body on different occasions (i.e., alternating hemi–grand mal, unilateral seizures, hemiclonic convulsions). The tendency to suffer temperature-sensitive seizures seems to persist over many years. In the series of Dravet et al. (33), short, single myoclonias were noted by some parents before onset of febrile seizures. Even though approximately 70% of cases begin with generalized or unilateral febrile seizures, focal seizures may occur already early in the course, but this is not the rule. Febrile seizures typically occur repeatedly within the first year of life and are frequently the dominating manifestation of the epilepsy in the first 9 months of age. Later, afebrile generalized seizures add to the febrile seizures and are soon followed by myoclonic seizures and atypical absences (33).
Dravet et al. (33) define “steady-state seizures” as those that prevail in many cases throughout the course. The authors recognize 10 different seizure types:
1. “GTCS” are basically identical to those encountered in idiopathic generalized epilepsy syndromes.
2. “Hemiclonic convulsions” are frequent in the first 3 years of life, and then they become rare. Postictal hemiparesis is a frequent feature. If they—by chance—reoccur on a specific side of the body, they may falsely be taken for focal seizures.
3. “Falsely generalized seizures” clinically appear like GTCS. On polygraphic recordings, this seizure can be resolved as bilateral asymmetric tonic contractions of different muscle groups.
4. “Unstable seizures” are clinically related to “falsely generalized seizures” with concomitant focal EEG discharges that change their origin between and during seizures.
5. Myoclonic seizures predominantly affect the axial muscles and may range from mild to severe, from simple head nodding to violent thrashes involving the entire body. Severe myoclonic seizures may result in falls and injuries. Repeated twitching of the head is a third observed type. These seizures may precede generalized tonic–clonic convulsions. Interictal myoclonias (without concomitant epileptic discharges on surface EEG) are a frequent feature, mainly observed in periods with high seizure frequency (roughly 70% of cases). Myoclonic seizures may abate over time.
6. Atypical absences may appear at any age during course; however, mostly after the first year of life. In our view, absences are not always “atypical” because regular 3-Hz spike slow-wave absences may be recorded. Absences may range from pure impairment of consciousness to absences with intermixed myoclonic seizures. Duration varies between 3 and 10 seconds in most cases.
7. Simple and complex focal seizures, frequently associated with strong autonomic reactions such as pallor, cyanosis, and sweating, are detectable in about two-thirds of cases. They may start already within the first year of life, but usually begin later. Adversive seizures and clonic seizures frequently in combination are typical manifestations.
8. Tonic seizures are rare in this syndrome. They seem to resemble those seen in Lennox–Gastaut syndrome, often with a myoclonic component.
9. Obtundation states are episodes of reduced attention (drowsy states). They occur in more than one-third of children. Usually, they are associated with erratic myoclonias involving the limbs or the face. These drowsy states may continue for hours or even days. In the EEG, dysrhythmic slow waves intermingled with spikes and sharp waves are characteristic. This state may evolve from an overt seizure or end in one.
10. A status of generalized tonic–clonic convulsions may occur without warning. Frequently fever, infection, or even photic stimulation may act provocative. If the status is then falsely treated by phenytoin (a sodium channel blocker), it may have an unfavorable outcome.
Besides intractable epilepsy, a variable degree of developmental delay (usually severe) characterizes the course. Children frequently develop an ill-defined moderately severe ataxia (60% to 70%) and mild pyramidal signs. Usually, ataxia is not disabling, will not prevent from walking, and will attenuate over the years. Besides fever, infection, and hot weather conditions, seizures may also be triggered by (hot) water immersion, joyful mood (e.g., birthday party), or physical exercise. Hyperkinetic behavior, especially at times of high seizure frequency, and autistic features are frequent findings. In general, the more severe the epilepsy, the more marked will be the developmental and behavioral problems. Death is reported in about 10% of larger historic series. Causes were mixed, ranging from status epilepticus to drowning, sudden unexplained death in epilepsy, and accidents (33).
EEG
In the first 1 or 2 years of life, the interictal EEG is frequently normal. Photosensitivity may be found in about 40% of cases. Over time, the background activity deteriorates. As reported by Doose, a rhythmic theta activity with accentuation over the central channels and independent of vigilance develops (20) (Fig. 19.3). Generalized regular and irregular spike–waves as well as multifocal spikes and sharp waves may evolve during the course. In unilateral seizures, lateralized spike–wave or slow spike–wave activity with intermittent irregular spike–wave is observed. In “falsely generalized seizures,” an initial amplitude reduction and spike–wave and slow spike–wave activity with changing asymmetry are observed. In unstable seizures, the epileptic EEG activity is similar but migrates from one brain region to another during the same seizure. In myoclonic seizures, spike–wave and polyspike–wave discharges occur simultaneously with the myoclonias. Absences are accompanied by irregular 2.5 to 3.5 generalized spike–wave discharges lasting mostly 3 to 10 seconds. Obtundation states (nonconvulsive status epilepticus) are characterized by generalized spike–wave and slow spike–wave discharges with intermixed fast and slow activities (30).
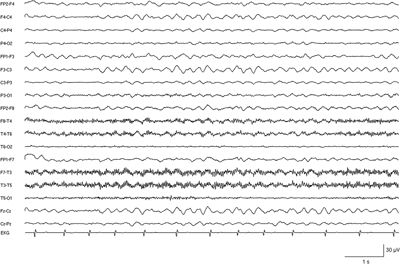
Figure 19.3. EEG recording in a severely retarded 14-year-old girl with SMEI showing persistent rhythmic slowing of background activity.
Treatment and Prognosis
Maybe the most important therapy option is to avoid provocative AED. In most cases of Dravet syndrome, SCN1A, the major sodium channel of inhibitory interneurons is reduced in activity or function to as low as 50% of normal. Application of sodium channel blockers (e.g., carbamazepine, oxcarbazepine, phenytoin, lamotrigine) may further aggravate this defect, resulting in seizure provocation up to status epilepticus. Head-to-head studies are impossible to conduct; however, retrospective analyses and clinical observation show that several agents are effective. Frequently, in the first 2 years of life, valproic acid is commenced. The next step would be to add either clobazam or topiramate, or successively both (35). If generalized tonic–clonic (especially fever- or infection-triggered) seizures and status still prevail, bromides (potassium bromide) may be of great help (22). From our point of view, bromides are possibly the most powerful drugs available for children with Dravet syndrome. Its potency in this syndrome should not be underestimated, but however, bromides predominantly control only GTCS. Children already treated with valproate and clobazam had a 70% seizure reduction under added stiripentol. The reduction of clonic and tonic–clonic seizures was most marked (36). Other drugs used with partial success are zonisamide, phenobarbital, and chloral hydrate. In addition, the ketogenic diet was reported to be successful (37,38). In children with a severe course, implantation of a permanent IV line (e.g., Port-a-Cath) is helpful to prevent or shorten repeated status epilepticus by rapidly administering phenobarbital and benzodiazepines.
Prognosis is dismal in basically all patients who bear the diagnosis of Dravet syndrome by right. Developmental delay usually becomes evident during the second or third year of life. Complete seizure freedom is not a realistic option. However, in some cases, reasonable results may be obtained by antiepileptic (combination) therapy. There are cases in the borderland (SMEB) who come closer to the clinical spectrum of GEFS+ and who may respond better to therapy.
Genetics and Molecular Diagnostics
Family history was formerly reported to be frequently positive for febrile convulsions and idiopathic epilepsy syndromes. However, a recent study could not reproduce these findings (39). By far, the most cases of Dravet syndrome are caused by defects in the SCN1A gene. In cases diagnosed by applying strict inclusion criteria (SMEI), heterozygous SCN1A mutations may be detected in up to 80% of cases (40). Other genes like SCN1B, SCN2A, and GABRG2 were detected in single patients or families with Dravet syndrome, but quantitatively do not play a significant role. About 95% of SCN1A mutations detected in Dravet syndrome patients appear de novo. The remaining 5% that are inherited are usually connected with milder epilepsy phenotypes resembling the GEFS+ spectrum. This is consistent with a mostly negative family history. SCN1A mutations are distributed over the entire gene. Truncating mutations are found in about 50% of SMEI patients. The remaining are mostly missense mutations loosely clustering at the ion pore positions of the channel protein. Splice site mutations and heterozygous deletions ranging from single exons to the entire gene are rare. Borderland patients (SMEB) also frequently show SCN1A mutations, but to a lesser degree (60% to 70%).
The spectrum of epilepsies associated with the SCN1A gene, however, is broader than Dravet syndrome (SMEI and SMEB) and GEFS+. It also covers some less well-defined infantile epileptic encephalopathies. All of them start within the first year of life and are therapy resistant. These are denoted “cryptogenic generalized epilepsy,” “cryptogenic focal epilepsy,” and “severe infantile multifocal epilepsy” (40).
Selection criteria to maximize chances of a SCN1A mutation detection are given in Table 19.2 (31). If four or more criteria are fulfilled, detection chances are about 70% or higher, using a combination of DNA sequencing and exon quantification assay (e.g., MLPA (multiplex ligation dependent probe amplification)).
Table 19.2 Common Features Recognized in Children with SCN1A Mutations that may be Used as a Guidance in Order to Estimate Chances of Detection in Mutational Analysis

In girls with Dravet (like) syndrome, PCDH19 defects are an important differential diagnosis and may be detected in about 15% of SCN1A-negative females. Juberg and Hellman (41) reported in 1971 on a syndrome named “epilepsy and mental retardation limited to females.” The publication was not really reflected for many years, because it reported a by then mostly unknown exceptional mode of inheritance, namely “X-linked dominant with male sparing.” Later the syndrome was rediscovered and defined in more detail by Dibbens et al. (42). Salient features are (41–43):
1. Only females affected
2. Epilepsy as a frequent phenotype (90%)
3. Seizure onset mostly in the second year of life (6 to 36 months)
4. GTCS, clonic seizures, myoclonic seizures, absences, unilateral seizures, focal seizures, febrile seizures
5. Seizures may remit spontaneously (mean 12 years)
6. Low intelligence in about 70% of cases
7. Developmental regression in about 50% of cases
8. Behavioral comorbidities are frequent (obsessive–compulsive, attention deficits, etc.)
9. Several reports of sudden unexpected death in epilepsy
10. No specific EEG pattern
PROGRESSIVE MYOCLONUS EPILEPSIES
Unverricht–Lundborg disease, Lafora disease, myoclonic epilepsy with ragged red fibers (MERRF), NCL, sialidosis, and dentato-rubro-pallido-luysian atrophy (DRPLA) are the archetypes of this disease group. Other disorders with variable phenotypes may—in some of the affected—take the course of a PME (see Table 19.1). PME are rare and comprise <1% of epilepsies. In their early course, some of them may be difficult to differentiate from idiopathic generalized epilepsies. Precise personal and family history and a thorough clinical and neurologic examination are pertinent to obtain diagnostic clues at an early stage (44). Extremely enlarged somatosensory evoked (Fig. 19.4) or visually evoked potentials induced by flashlight, an enhanced long-latency reflex in response to electric stimuli referred to as C-reflex, and an abnormal reaction to paired pulse transcranial magnetic stimulation all reflecting increased cortical excitation or decreased cortical inhibition may be diagnostically helpful neurophysiologic findings. Examination of the eye background is also important, since this may reveal optic atrophy, a cherry red spot or retinitis pigmentosa.
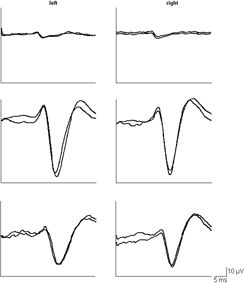
Figure 19.4. Somatosensory evoked potentials (SEPs) in a 13-year-old boy with progressive myoclonus epilepsy (PME) and in a healthy male of same age (top). Giant SEPs are recorded in the patient with PME reflecting extreme cortical hyperexitability to sensory stimuli (middle). Notice the substantial reduction of the SEP amplitude during treatment with levetiracetam (bottom).
Related posts:
 Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


