Progressive Myoclonus Epilepsies
Samuel F. Berkovic
Introduction
Progressive myoclonus epilepsy (PME) is an uncommon epilepsy syndrome caused by a large number of rare specific disorders. In its fully developed form with florid, unremitting myoclonic seizures and progressive neurologic deterioration, the syndrome can hardly be missed. Diagnosis of the PME syndrome can be more difficult in the early stages, and confusion with more benign epilepsies is common. Diagnosis of the specific type of PME is challenging, as most individual clinicians’ experience with these rare disorders is limited. Molecular genetics has had an enormous impact on the clinical approach to these disorders, and PME is the clinical syndrome par excellence showing the value of careful clinicomolecular correlations leading to advances in practical clinical diagnosis and fundamental biologic understanding. This chapter will focus on diagnosis of the PME syndrome and of the more common specific causes.
Historical Perspectives
The rarity and complexity of the disorders causing PME have resulted in a confusing literature since the first description by Unverricht in 1891 (Fig. 1).130 Eponymous names were used in conflicting ways, and there were often erroneous putative clinicopathologic correlations. In particular, the term Ramsay Hunt syndrome generated enormous confusion (for review see references 11, 18, 84, and 95).
Pathologic studies since the 1930s established that there were at least three separate pathologic substrates of the PME syndrome: Lafora bodies, lipid storage, and “degenerative” changes.36,54,56 However, in clinical practice, PME was often the final clinical diagnosis, as there was no way to diagnose specific forms during life.
Over the last three decades a number of clinical, pathologic, genetic, biochemical, and molecular advances have led to considerable clarification of this subject, allowing a sophisticated and rational approach to diagnosis in life of the patient with PME.7,14,46,84,116 First, in many of the specific causes of PME, characteristic clinical patterns have been recognized. Second, ethnic and geographic clusters of certain disorders have been identified, accounting for the different perspective of PME by authors in various countries. Third, the broad pathologic group of “lipidoses” causing PME has been classified by clinical, biochemical, and pathologic studies into neuronal ceroid lipofuscinoses, sialidoses, and Gaucher disease. Fourth, the mitochondrial disorder MERRF (myoclonus epilepsy and ragged red fiber syndrome) was discovered and found to be a major cause of PME. Fifth, the “degenerative” type of PME was found to be heterogeneous, comprising at least three distinct conditions: Unverricht-Lundborg disease, MERRF, and dentatorubral-pallidoluysian atrophy. Sixth, minimally invasive methods for diagnosis during life were developed. Finally, important molecular genetic findings have further refined understanding of these disorders and are playing an increasing role in routine diagnosis.
Definitions
The syndrome of PME consists of myoclonic seizures, tonic–clonic seizures, and progressive neurologic dysfunction, particularly ataxia and dementia. Onset can be at any age, but is usually in late childhood or adolescence. There are a large number of causes of the PME syndrome; most are due to specific genetic disorders, which can now be accurately diagnosed in life.7,46
Myoclonus in PME is typically fragmentary and multifocal, and often precipitated by posture, action, or external stimuli such as light, sound, or touch. It is particularly apparent in facial and distal limb musculature. Bilateral massive myoclonic jerks, which tend to involve proximal limb muscles, may also occur.
The origin of and generators for myoclonus in PME is a confusing and controversial area. Neurophysiologic studies show that some but not all myoclonic jerks are accompanied by obvious electroencephalographic (EEG) spikes, polyspikes, or spike-and-wave complexes. Where EEG accompaniments of jerks are not obvious, back-averaging techniques may reveal preceding EEG changes. These data, coupled with the frequent finding of giant somatosensory-evoked potentials (SSEPs) and of evidence suggesting abnormal cortical hyperexcitability using transcranial magnetic stimulation, suggest that the myoclonic jerks are often of cortical origin—cortical reflex myoclonus. Electrophysiologic studies of bilateral jerks suggest that some are generated in the cortex unilaterally and spread rapidly contralaterally via the corpus callosum, whereas others may be generated in the brainstem—reticular reflex myoclonus.50,109,118,119 The lack of demonstrable EEG change with some jerks might lead to the conclusion that these are examples of “nonepileptic” myoclonus, akin to that seen in certain movement disorders. Unfortunately, no absolute clinical or indeed experimental technique exists to determine the epileptic nature or otherwise of particular jerks. Moreover, the coexistence of “epileptic” and “nonepileptic” myoclonus in one patient is counterintuitive. From a pragmatic clinical viewpoint, therefore, “epileptic” myoclonus can be diagnosed in cases where tonic–clonic or other seizures coexist or where obvious epileptiform discharges accompany some, but not necessarily all, myoclonic jerks.
 FIGURE 1. Heinrich Unverricht of Magdeberg (1853–1912). First clinical description of progressive myoclonus epilepsy in 1891. (Reproduced from Berkovic SF, Andermann F. The progressive myoclonus epilepsies. In: Pedley TA, Meldrum BS, eds. Recent advances in Epilepsy. Edinburgh: Churchill Livingstone; 1986:157–187.) |
Epidemiology
PMEs account for <1% of people with epilepsy seen at specialist centers.46 Series from different countries reveal considerable
geographic and ethnic variability in the occurrence of specific types of PMEs.1,36,40,46 Details of known geographic clusters of specific PMEs are given below. Knowledge of the patient’s ethnic background can provide an essential clue to the likely differential diagnosis of the type of PME.
geographic and ethnic variability in the occurrence of specific types of PMEs.1,36,40,46 Details of known geographic clusters of specific PMEs are given below. Knowledge of the patient’s ethnic background can provide an essential clue to the likely differential diagnosis of the type of PME.
The incidence and prevalence of specific PMEs are largely unknown. In Finland, Unverricht-Lundborg disease has an incidence of at least 1 per 20,000,95 but outside the Baltic region the incidence is probably at least an order of magnitude less, although a recent study in The Netherlands using molecular methods suggested that Unverricht-Lundborg disease is underdiagnosed.35
Specific Disorders
Unverricht-Lundborg Disease
Etiology and Basic Mechanisms
Unverricht-Lundborg disease is the prototypic cause of PME.65,77,130 No storage material is present but there is neuronal loss and gliosis particularly affecting the cerebellum, medial thalamus, and spinal cord.51
It is an autosomal recessive condition95 initially recognized as a geographic cluster in Finland and eastern Sweden (Baltic myoclonus). An erroneous, but frequently held, view is that this disorder is confined to the Baltic region. Clusters of a phenotypically identical disorder occur in Southern Europe and North Africa, so-called “Mediterranean myoclonus.”47 It is also found sporadically worldwide in Caucasians, Blacks, and Japanese.40,84 Indeed, it appears that Unverricht’s original family was of Baltic German extraction, and not Estonian as widely believed.
The disorder was linked to the long arm of chromosome 21 in Finnish cases in 199171 and cystatin B was identified as the responsible gene in 1996.104 The clinical prediction that similar cases seen outside the Baltic region have the same condition was confirmed by showing the identification of mutations in cystatin B (CSTB) in families from around the world. The most common mutation, responsible for about 90% of abnormal alleles, is an unstable expansion of a dodecamer repeat in the 5′ untranslated promoter region. The remaining mutations are missense mutations.69,70,72 CSTB is a cysteine protease inhibitor. The CSTB mutations lead to marked reduced expression of CSTB mRNA. Development of a mouse model with targeted disruption of the mouse Cstb gene has shown increased apoptosis affecting particularly cerebellar granule cells. It has been suggested that deficiency of CSTB protein results in increased activity of cathepsins with increased apoptosis in specific neuronal cell types.72,103,117
A variant has been described where CTSB has been excluded as the causative gene. This form maps to chromosome 12 but the gene is presently unknown.15
Clinical Presentation
Clinical onset is with myoclonus or tonic–clonic seizures between the ages of 8 and 13 (mean 10, range 6 to 16 years). The myoclonus is usually quite severe and may be precipitated by movement, stress, and sensory stimuli. Repetitive morning myoclonus is also typical, frequently building up and culminating in a major tonic–clonic seizure.63,64 Seizures may be difficult to control, but progression in terms of ataxia and dementia is mild and late. The clinical course is variable and there may be considerable intrafamily variation in the
severity of the seizures. Some patients are relatively mildly affected and survive to old age.78 A more fulminant course with death within a few years of onset has been observed; this outcome is rarely if ever seen now and may have been due to unrecognized deleterious effects of phenytoin.40,58
severity of the seizures. Some patients are relatively mildly affected and survive to old age.78 A more fulminant course with death within a few years of onset has been observed; this outcome is rarely if ever seen now and may have been due to unrecognized deleterious effects of phenytoin.40,58
The EEG background may show some diffuse theta that increases over years, as well as some frontal beta activity. Epileptic activity comprises 3- to 5-Hz spike-wave or multiple spike-wave activity with the maximum field being anterior. Sporadic focal spikes, particularly in the occipital region, may be seen but are usually not prominent (Fig. 2). Photosensitivity is typically marked. During non–rapid eye movement (REM) sleep the spike-wave activity is diminished.18,64
Diagnostic Evaluation
Unverricht-Lundborg disease is recognized clinically by its characteristic age of onset and clinical pattern, with an absence of other clinical or pathologic features. Diagnosis is confirmed by molecular analysis of the cystatin B gene.
Myoclonus Epilepsy and Ragged Red Fibers
Etiology and Basic Mechanisms
The MERRF syndrome has emerged as one of the most common causes of PME. It may be familial or sporadic. Most familial cases of MERRF are transmitted through the maternal line and are examples of mitochondrial inheritance.111 The peculiarities of mitochondrial inheritance provide an explanation for the wide phenotypic variability in patients with MERRF and for the extraordinary intrafamily variation.
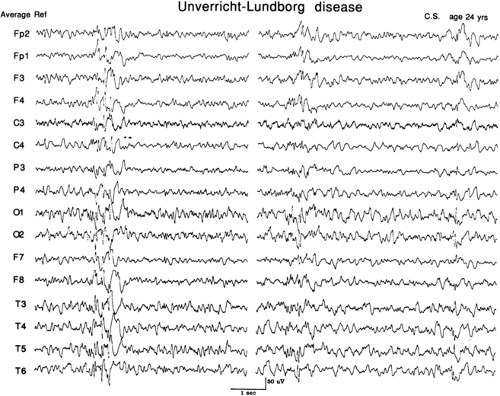 FIGURE 2. Unverricht-Lundborg disease. Waking electroencephalogram in a 24-year-old woman showing generalized polyspike-and-wave discharges on a slow background (left panel) with occasional focal occipital discharges (right panel). |
A single base substitution at nucleotide pair 8344 of mitochondrial DNA, causing an A-to-G substitution in the tRNALys gene, occurs in many familial cases of MERRF.120 The fact that this mutation affects tRNA, rather than a gene for a respiratory enzyme, probably explains the heterogeneous results for respiratory enzyme assays reported in MERRF. This tRNALys mutation appears to underlie most, but not all, familial cases and some sporadic examples of MERRF.17,52,53,138 Other rare identified molecular causes of MERRF also affect the tRNALys gene.121 Recently, autosomal recessive mutations in the nuclear encoded mitochondrial gene polymerase-γ (POLG) have been identified in some MERRF cases.129
Pathologically the brain shows “degenerative” changes, particularly affecting the dentate nucleus and inferior olive. In more severely affected cases, lesions typical of Leigh disease are also found. Positron emission tomography shows decreased metabolism for glucose and oxygen with relatively preserved cerebral blood flow, findings compatible with a respiratory chain defect.13 Phosphorus magnetic resonance spectroscopy of the brain is normal but studies of resting muscle show an increase of inorganic phosphate and a decrease of the phosphocreatine-to-inorganic phosphate concentration ratio.86
Clinical Presentation
MERRF was first described in cases with a florid clinical myopathy and myoclonus epilepsy.43,128 It is now clear that the clinical spectrum of MERRF is extremely broad. It should be suspected in a wide variety of situations, even when clinical and pathologic evidence of myopathy is absent.8,13 Symptoms can begin at any age and there may be marked intrafamily variation in the age of onset and clinical severity.13,111 The clinical features include myoclonus, tonic–clonic seizures, dementia, ataxia, and less common findings of myopathy, neuropathy, deafness, and optic atrophy. Some cases show striking axial lipomas. Occasional patients or families have focal neurologic events and there is an overlap with the syndrome of mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS), where strokelike episodes frequently preceded by migrainous headaches with vomiting are characteristic.8,102
It has been previously suggested that a wide variety of cases of PME known by eponyms, clinical signs, or particular patterns of system degeneration were examples of mitochondrial disease.4,7,13 This has now been confirmed for PME with lipomas,13,39 and for at least some cases of PME and deafness87,131 and of so-called Ramsay Hunt syndrome.4,10,84 PME with Friedreich ataxia122 and PME with deafness, focal cerebral deficits, alopecia, and a transient response to biotin21 may also be due to mitochondrial disease.
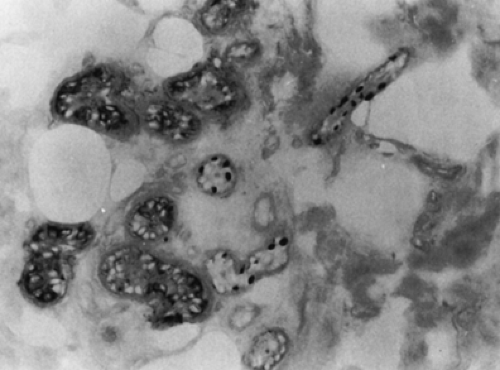 FIGURE 3. Lafora disease, skin biopsy. Cryostat section of skin stained with periodic acid-Schiff showing oval densely staining inclusions in eccrine duct cells. The secretory ascini (on the left) show normal glycogen staining (×225). (Reproduced from Berkovic SF, Andermann F. The progressive myoclonus epilepsies. In: Pedley TA, Meldrum BS, eds. Recent advances in Epilepsy. Edinburgh: Churchill Livingstone; 1986:157–187.) |
The EEG shows slowly progressive background slowing paralleling degree of clinical deterioration. There are generalized spike-and-wave discharges at 2 to 5 Hz or multiple spike-and-wave discharges. Sporadic occipital spikes and sharp
waves may be seen. Prominent photosensitivity may occur. Non-REM sleep is disorganized and spike-and-wave discharges are diminished.18,123
waves may be seen. Prominent photosensitivity may occur. Non-REM sleep is disorganized and spike-and-wave discharges are diminished.18,123
Diagnostic Evaluation
The unifying feature of these cases is dysfunction in the mitochondrial respiratory chain. This is most simply demonstrated by ragged red fibers in skeletal muscle, although these can be absent. Biochemical assays of the mitochondrial respiratory enzymes may show abnormalities, but these too may be normal.13,134
Diagnosis can usually be suspected clinically and may be difficult to confirm with laboratory markers. The clinical clues to the diagnosis include deafness, optic atrophy, myopathy, lipomas, intrafamily variation in age of onset and severity, and a pattern of inheritance compatible with maternal transmission.13 Serum lactate, ragged red fibers, and respiratory enzyme activities in muscle can all be normal in patients known to be affected (e.g., family members of proven cases). Magnetic resonance spectroscopy of muscle may show elevated levels of inorganic phosphate and a decrease of the phosphocreatine-to-inorganic phosphate concentration ratio.86 Molecular defects in mitochondrial DNA or POLG can be detected, when present, in peripheral blood or muscle.53,138 Screening for the mitochondrial DNA 8344 mutation should be done first; if negative, then more extensive DNA testing of mitochondrial DNA or POLG may be indicated.
Lafora Disease
Etiology and Basic Mechanisms
Lafora disease is characterized by the presence of Lafora bodies, which are polyglucosan inclusions found in neurons and in a variety of other sites including heart, skeletal muscle, liver, and sweat gland duct cells25,68 (Fig. 3).
It is an autosomal recessive condition. The largest series have been reported from Southern Europe,125 but it is found worldwide, apparently without a marked racial or ethnic predilection. Approximately 90% of cases have mutations in the gene EPM2A, which encodes a dual phosphatase known as laforin,89,114 or in EPM2B (also called NHLRC1), which codes for an E3 ubiquitin ligase known as malin.28,48 There is evidence for a third as yet unknown locus.27
Clinical Presentation
Onset is between the ages of 10 and 18 years with a mean age of onset of 14. Clinical features are myoclonus, tonic–clonic seizures, and relentless cognitive decline. Focal seizures, particularly arising from the occipital regions, occur in about half the patients. Recognition of Lafora disease in its fully developed form is not difficult. At the onset, however, the disorder can resemble a typical benign adolescent generalized epilepsy with no evidence of cognitive decline. It may also present as a dementing illness with relatively infrequent seizures, or it may mimic a nonspecific secondary generalized epilepsy because myoclonus is not obvious.11,107,110 The prognosis of Lafora disease is dismal, with death occurring 2 to 10 years after onset and the mean age of death being 20 years.
The clinical picture, including the relatively narrow age range of onset and relentlessly progressive course to death within 2 to 10 years of onset, is constant in all reports with the exception of a few cases. These cases, sometimes erroneously labelled as “type Lundborg,” had symptoms beginning in late adolescence or early adult life with a milder protracted course.36,60,66 Certain mutations in EPM2B may cause a milder course.6 Conversely, an early-onset form with marked cognitive decline has been reported with mutations in exon 1 of EPM2A.44
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


