INTRODUCTION
In 1963, J. Clifford Richardson (1) described an unusual syndrome of supranuclear gaze palsy, and progressive axial rigidity, pseudobulbar palsy, and mild dementia. Together with John Steele and Jerzy Olszewski (2), they reported extensive subcortical neurofibrillary degeneration predominantly in the globus pallidus, subthalamic nucleus, substantia nigra, and dentate nucleus, which were eventually used to establish the new clinicopathologic entity of progressive supranuclear palsy (PSP), also known as Steele–Richardson–Olszewski syndrome. There are a number of distinct clinical syndromes relating to pathologic PSP, and the classic clinical presentation has now been renamed Richardson’s syndrome (RS, or PSP-RS). PSP-RS and clinical variants all share similar neuropathologic features, and fulfill the neuropathologic criteria for PSP but can be separated by differences in both their clinical features and the regional severity of pathology (3). The current operational criteria are only limited to the clinical diagnosis of PSP-RS (4,5) (Table 14.1), and no accepted guidelines for the clinical diagnosis of PSP subtypes have been compiled. With the lack of reliable diagnostic biomarkers for PSP, accurate diagnosis depends on clinical acumen and the recognition of different clinical presentations.
EPIDEMIOLOGY
The prevalence of PSP in the United Kingdom has been estimated at 6.4 and 6.5 per 100,000 in active case-finding studies in Newcastle and London (6,7). A study in Japan reported a similar prevalence of 5.8/100,000 (8). A series from the Queen Square Brain Bank for Neurological Disorders of 103 pathologically confirmed cases of PSP, of which 63% were men, reported that the mean age of onset of the neurologic symptoms was 66.4 (± 12) years, age at death was 73.5 (± 7.5) years, and disease duration was 7.0 (± 3.7) years (9). Other clinical subtypes such as PSP–parkinsonism (PSP-P) and PSP–pure akinesia with gait freezing (PSP-PAGF) have a longer disease duration when compared to PSP-RS (3).
RISK FACTORS
Advanced age is the only established risk factor for PSP, although case–control analyses are difficult to carry out because of the rarity of the disease. An association between PSP and low likelihood of completing at least 12 years of education was identified in one study (10). No relationship between PSP and smoking or head injury has been identified. On the Caribbean Island of Guadeloupe in the French Antilles, there is a high prevalence of cases with atypical parkinsonism predominantly affecting the African-Caribbean population, of which a third has a PSP-like syndrome. The Guadeloupean atypical parkinsonism may be associated with the traditional consumption of soursop, which contains the mitochondrial complex I inhibitor, annonacin (11). The relationship between the Guadeloupian disease and PSP-RS remains uncertain.
CLINICAL FEATURES
RICHARDSON’S SYNDROME
Patients, presenting in their late 50s or 60s, may complain of visual symptoms, dizziness, clumsiness, unsteadiness, unprovoked falls, difficulties in walking, or fatigue, whereas family members frequently comment on irritability, apathy, introversion, depression, and softening of speech. A fifth of patients with PSP had predominant behavioral and cognitive features at presentation similar to frontotemporal dementia, without preceding motor features (12). Delay in diagnosis of 3 to 4 years is common (6,13). As the disease progresses, postural instability and falls are the most characteristic and disabling features (14). Fractures and soft tissue injuries are common (15). The gait is slightly broad based, which may appear ataxic, resembling that of a drunken sailor or a dancing bear, and may be inappropriately brisk with a propensity to fall backward. Early in the disease course, the gait may appear relatively normal in the clinic, despite a history of frequent falls and injuries, and gradually, there may be gait ignition failure and freezing of gait. Patients often pivot on one foot when turning, which further compromises their balance. Axial rigidity leads to an upright extended posture and sometimes retrocollis. On rising from a chair, the patient typically extends the trunk and neck and may spontaneously topple backward. Motor recklessness resulting from frontal lobe dysfunction may lead to falls and injuries on attempting to rise from a sitting position rapidly, a feature known as “the rocket sign.” Difficulty with feeding is common, caused by a combination of vertical gaze palsy, reduced dexterity, overfeeding, and swallowing difficulties. An astonished and worried facial expression relates to focal dystonia of the procerus muscle (procerus sign), frontalis overactivity, reduced eye blink, lid retraction (Collier’s sign) (16), gaze palsy, and spasticity, resulting in a deeply lined facial contour. The distinctive “Mona Lisa fixity of gaze” along with the typical gait and posture permit the clinical suspicion of PSP as the patient enters the consultation room.
Fine finger movements are slow and clumsy. Repetitive finger tapping is small in amplitude (hypokinesia) with good speed and without decrement (17). Handwriting is untidy and small, and a characteristic “fast micrographia,” may occur particularly in the PSP subtype, PSP-PAGF (17,18).
Orthostatic hypotension is not a feature of PSP, unlike multiple system atrophy (MSA). Urinary urgency, retention and incontinence, constipation, and erectile dysfunction are, however, common as the disease progresses. Polysomnographic evaluation of 20 patients with PSP revealed marked sleep abnormalities, including an overall lower sleep efficiency, rapid eye movement (REM) sleep without atonia in 85% of patients and REM sleep behavior disorder in 35% (19). Sleep problems were correlated with disease severity and worsening dementia in one study (20).
| National Institutes of Neurological Disorders and Stroke–Society of Progressive Supranuclear Palsy Consensus Clinical Research Diagnostic Criteria (4) |
Clinically “possible” PSP | Clinically “probable” PSP |
All three of these: Gradually progressive disorder Onset at age 40 or later No evidence for competing diagnostic possibilities Plus either of these: Vertical gaze palsy Slowing of vertical saccades and prominent postural instability with falls in the first year | All five of these: Gradually progressive disorder Onset at age 40 or later No evidence for competing diagnostic possibilities Vertical gaze palsy Slowing of vertical saccades and prominent postural instability with falls in the first year |
Criteria that would exclude the diagnosis of PSP | |
Recent encephalitis Alien limb syndrome, cortical sensory defects, or temporoparietal atrophy Psychosis unrelated to dopaminergic treatment Important cerebellar signs Important unexplained dysautonomia Severe, asymmetric parkinsonian signs Relevant structural abnormality of basal ganglia on neuroimaging Whipple’s disease on cerebrospinal fluid polymerase chain reaction | |
In the late stages of disease, a growling drawl, swallowing difficulties, and emotional lability contribute to the deterioration of quality of life. Drooling, coughing, choking, inspiratory sighs, and, occasionally, stereotyped moaning or groaning, occur as the illness advances. Pseudobulbar palsy with slow spastic tongue movements, reduced gag reflex, brisk jaw, and facial reflexes are common findings. Severe nuchal rigidity is disproportionate to the moderately increased tone in the limbs. Reflexes are usually brisk with a positive Babinski sign is observed in one-fifth of patients (9). Jerky postural tremor of the limbs (9), limb dystonia, or arm levitation is observed in a minority of patients with RS, but is more common in other PSP subtypes (21–23).
In the terminal phase, patients are unable to communicate due to anarthria, complete ophthalmoplegia, marked rigidity, and are confined to bed. Death frequently results from respiratory failure, aspiration pneumonia, urinary tract infection, or pulmonary embolism.
NEURO–OPHTHALMOLOGIC FEATURES
Blurred vision, difficulty focusing, dry eyes, photophobia, and double vision are frequent early complaints. Difficulties navigating a plate of food, stairs, or ceiling caused by slowness of vertical eye movements are reported by observant patients. As the disease advances, the ability to read is eventually lost because of the saccadic eye movement disorder.
| Eye Movement Abnormalities in PSP by Disease Stages |
Early stage | Hypometric saccades, particularly downward Disordered opticokinetic nystagmus, particularly downward Slowing of downward saccades Square-wave jerks |
Middle stage | Restriction of range of vertical pursuit eye movements, particularly downward Impaired convergence Disordered Bell’s phenomenon Visual grasping Lid retraction with reduced blink rate Apraxia of lid opening or closing |
Late stage | Restriction of range of voluntary horizontal gaze Loss of oculocephalic reflex Dysconjugate gaze Disabling blepharospasm |
Square-wave jerks, in which eyes oscillate horizontally across the midline during visual fixation, is an early sign (Table 14.2). It is not specific for PSP and is also frequently observed in MSA, cerebellar disorders, and occasionally in Parkinson’s disease (24). Impairment of convergence, defective pupillary responses with accommodation, and absence or slowing of the quick phase of optokinetic- and caloric-induced nystagmus are other early signs (25,26).
A supranuclear vertical gaze palsy or slowing of vertical saccades is necessary for a firm diagnosis of PSP (4). Saccadic eye movements are the most informative bedside assessments used to clinch the diagnosis. Saccades are tested by asking the patient to shift gaze between two fixed visual targets, such as examiner’s nose and pen, displaced horizontally or vertically. Following the command to shift gaze, the time taken to initiate saccade, the traveling speed of saccade, and the trajectory should be noted (27). In early PSP, before the range of vertical gaze is affected, slowing of vertical saccades, either up or down, is the key finding that suggests PSP (28). The trajectory of vertical saccades may be oblique or curved (“round the houses sign”) (29). The time to initiate horizontal saccades following command is normal in PSP but is typically delayed in corticobasal syndrome (CBS) (30). Voluntary gaze on command without a specific target (“look down” or “look up”) is in general more difficult than shifting gaze to a target, which is more difficulty than pursuit, tested by asking patient to pursue a moving target, and reflex gaze is the last to be affected. Bilateral impairment of the antisaccade task, which correlates with frontal lobe dysfunction, is another early sign (31). It is tested by instructing the patient to look in the direction opposite to the visual stimulus placed on a horizontal visual plane (“look away from examiner’s waving hand”).
As the disease advances, the range of vertical eye movements diminish, followed by horizontal eye movements, and eventually complete ophthalmoplegia ensues (32). Vestibulo–ocular reflexes (VOR), or “doll’s eye” maneuver, are almost always preserved until the terminal stages, but increasing neck rigidity may make the maneuver difficult to elicit (33). Preserved VOR indicates the gaze deficit is supranuclear. In the terminal stage, the loss of VOR is an occasional finding, suggesting nuclear involvement in the brain stem (34).
Visual difficulties in PSP are also caused by impairment in orienting attention (31). Patients tend not to look at the right direction when conversing or reaching out for objects. Visual attention downward is particularly impaired. Difficulties in inhibiting visual orientation leads to visual grasping, in which patients appear to be magnetically attracted to objects they have walked past with intermittent head movements and full eye deviation that are otherwise impossible on command (35). Sustained head twisting movements with eye deviation opposite to the direction the patient’s body can occur and are related to a disturbed head position resetting mechanism (36,37).
Spontaneous eyelid closure, may be caused by tonic inhibition of levator palpebrae, pretarsal blepharospasm, or apraxia of eyelid opening, is a common feature that can lead to functional blindness (38). Patients often resort to using sensory tricks such as lightly touching the corners of the eyes to temporarily alleviate the symptom. Blink rate is severely reduced to an average of 0 to 4 times per minute (38). Unlike in MSA, CBS, and Parkinson’s disease, electrical blink reflexes are severely impaired in PSP because of the profound pathology in the brain stem (39). Bell’s phenomenon is absent in PSP (26).
NEUROPSYCHOLOGICAL AND COGNITIVE FEATURES
Assessment of higher cortical function is useful in differentiating PSP from other parkinsonian conditions. Frontal lobe dysfunction is the most consistent deficit (40). Behavioral change, apathy, and difficulties in planning or judgment in everyday tasks may be early features. Disinhibition with emotional lability, such as uncontrollable laughter or crying, aggressive outbursts, and inappropriate sexual behavior can be disabling features. Compulsive behavior is occasionally prominent. Executive dysfunction, including difficulties with shifting mental set, sorting, problem-solving, abstract thinking, motor sequencing, and lexical fluency, is revealed in the clinical history and detailed neuropsychometric testing. The frontal assessment battery (FAB) is a short but useful bedside quantification of the frontal lobe function (41). A score of below 12 (out of 18) has a sensitivity of 77% and specificity of 87% in differentiating frontal lobe dysfunction from amnestic dementia (42). Excessive dependence on environmental stimuli in PSP is another frontal lobe deficit specifically related to the lack of inhibition normally exerted by the prefrontal cortex. This can be demonstrated by the automated tendency to react to sensory cues, such as to imitate examiner’s movements (imitation behavior), to use object (utilization behavior) and to grasp objects put in front of them (forced grasping) in the absence of any explicit verbal orders. The Dementia Rating Scale (43) and Addenbrookes Cognitive Examination (ACE) (44) are other useful clinimetric scales to differentiate PSP from CBS, MSA, and Alzheimer’s disease by the finding of disproportionate impairments of different cognitive domains. Initial letter verbal fluency is a brief but very helpful bedside test to identify frontal cognitive impairment and distinguish PSP from Parkinson’s disease—PSP patients can only generate seven initial letter words or less in 1 minute (44).
NATURAL HISTORY
The National Institute of Neurological Disorders and Stroke (NINDS) PSP clinical diagnostic criteria included “falls within the first year” as a prerequisite for the diagnosis of “probable” PSP (4) (see Table 14.1). However, a significant proportion of patients with PSP start to fall after 2 years of disease onset, and a United Kingdom Brain Bank series reported that the average period to the first fall in PSP was 16.8 months (15). The NNIPPS (Neuroprotection and Natural History in Parkinson Plus Syndromes) study found that falls within the first year was present in only 48.6% of PSP patients and were also observed in 21.9% of the MSA group. PSP has a faster rate of progression in terms of functional disability and survival compared to MSA (45). Another study used Kaplan–Meier survival curves to determine the average intervals to seven clinical milestones representative of disease progression in 110 patients with pathologically confirmed PSP, using retrospective data (46). Intervals from disease onset to the development of frequent falls was 3.9 (±2.5) years, cognitive impairment 4.2 (±2.9) years, unintelligible speech 6 (±2.5) years, residential care was 6.1 (±3.0) years, urinary catheter 6.3 (±3.1) years, wheelchair dependence 6.4 (±2.7) years, and severe dysphagia 6.4 (±2.4) years. A shorter survival was associated with male gender, older age of disease onset, and shorter interval to reach the first milestone (46). Falls, dysarthria, diplopia within 1 year, or dysphagia within 2 years were also linked with a worse prognosis as shown in an earlier study (7). A PSP rating scale comprised of 28 items in six categories, including daily activities, behavior, bulbar, oculomotor, limb motor, and gait, provides useful quantitative assessment in clinical practice and research trials (47). The scores, ranged from 0 to 100, correlate well with subsequent survival, and the mean progression rate is +11.3 (±11) points per year. The motor subscale of the Unified Parkinson’s Disease Rating Scale has been shown to reliably assess motor disability in PSP (48).
PSP SUBTYPES
In the seminal paper, Steele, Richardson, and Olszewski emphasized that none of the nine patients described were considered to have a clinical picture of parkinsonism in view of the absence of tremor, limb bradykinesia and rigidity, flexed posture, or typical parkinsonian gait (2). The authors drew a clear distinction between PSP and paralysis agitans but anticipated that further observations of new cases would broaden the clinical spectrum of this condition. Subsequent clinicopathologic studies indeed identified cases with PSP–tau pathology and atypical clinical features, leading to the recognition of different clinical subtypes. Cases with a specific clinical subtype are classified as such based on their predominant presenting clinical picture (Table 14.3). As the disease advances, overlap of clinical features or temporal evolution to a clinical picture similar to RS frequently occurs in the later stages.
PROGRESSIVE SUPRANUCLEAR PALSY–PARKINSONISM
Williams and colleagues used a data-driven approach to distinguish a subgroup of cases with predominant parkinsonism from the classic clinical picture of PSP (9). The parkinsonian subgroup, commonly misdiagnosed as having Parkinson’s disease, comprised of a third of the 103 cases evaluated in the study and were all confirmed to have PSP–tau pathology. Patients with PSP-P present with asymmetric limb bradykinesia and rigidity and do not have supranuclear vertical gaze palsy in the early stage. About 40% of patients have a jerky postural tremor or 4 to 6 Hz rest tremor. A moderate response to levodopa therapy is encountered in half of the patients, but the benefit is usually less than that in Parkinson’s disease and rarely sustained for more than a few years. The clinical picture of PSP-P is most distinct in the first 2 years, but as the disease advances, the clinical phenomenology gradually becomes more similar to RS (9). In the early stages, it can be very difficult to separate PSP-P from Parkinson’s disease, and early clinical pointers, including rapid progression, prominent axial symptom, and an attenuated response to levodopa, when present, can be useful (9). Small amplitude finger taps without decrement may be an early clinical clue, but longitudinal objective measurements of patients with the PSP-P subtype are needed (17). Later in the disease course, levodopa-induced dyskinesia, visual hallucinations, and autonomic dysfunction are more suggestive of Parkinson’s disease, and are usually not prominent in patients with PSP pathology (49). Falls and cognitive decline occur later in PSP-P than in RS and may explain an average survival of 9 years in PSP-P as compared to 6 years in RS (46).
| Clinical Features of PSP-RS, PSP-P, PSP-PAGF, PSP-CBS, PSP-PNFA, PSP-FTDbv, Parkinson’s Disease, and MSA-P |
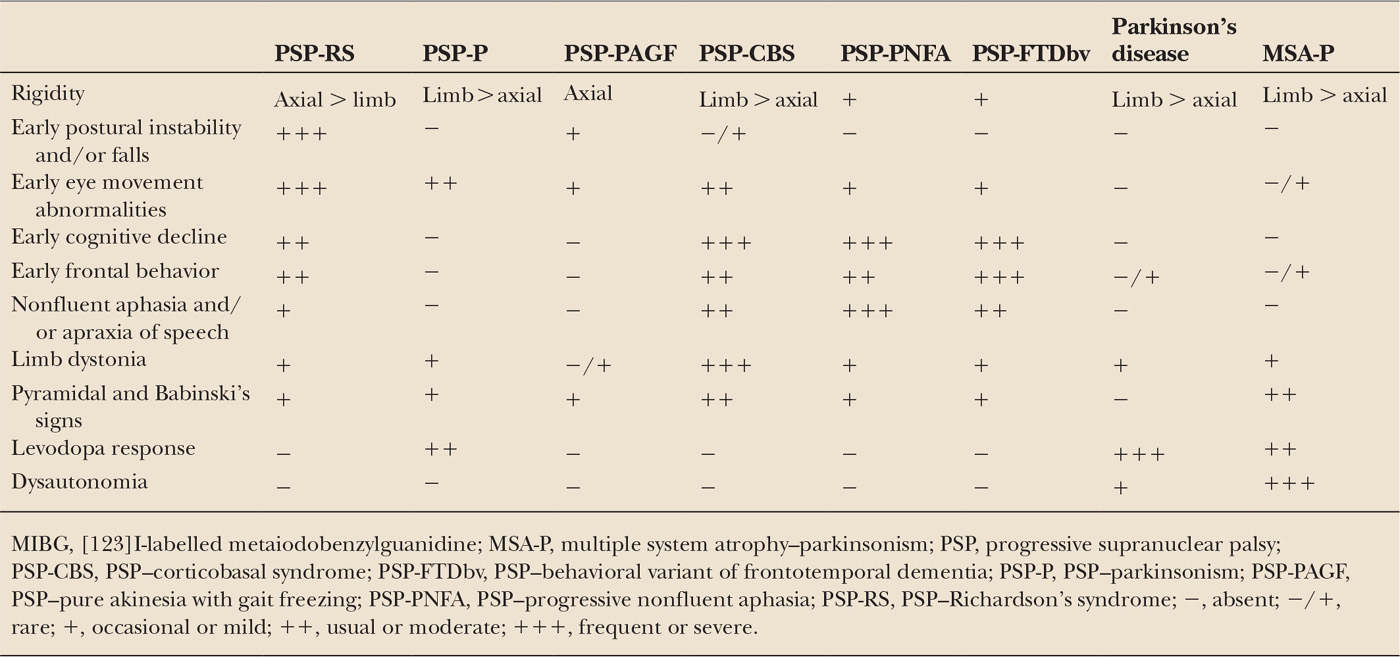
In clinical practice, some patients fall into the gray area between the distinct clinical pictures of PSP-P and RS. These patients have overlap of clinical features, such as asymmetric bradykinesia, rigidity and tremor as well as early postural instability, subtle eye movement abnormalities, and frontal subcortical deficits, and are diagnosed as having PSP-P because their parkinsonian symptoms are more conspicuous than those of RS. In a few patients, a pure Parkinson’s disease–like syndrome predominates until death and eye movement abnormalities never appear (9,50). A sustained levodopa response, levodopa-induced choreiform dyskinesia, and long disease duration characterize this distinct form of PSP-P, which represents the far end of the clinical spectrum and is associated with more restricted distribution of tau pathology (9). Further prospective studies are needed to define the full clinical phenotype of PSP-P.
PROGRESSIVE SUPRANUCLEAR PALSY–PURE AKINESIA WITH GAIT FREEZING
Pure akinesia was first described in 1974 in two Japanese patients (51), and the clinical syndrome was later considered as a variant of PSP (52,53). PSP-PAGF is characterized by pronounced gait ignition failure and start hesitation, often as the isolated clinical features for several years (54). Hypophonia, facial hypomimia, and fast micrographia may be early associated symptoms. Subsequently, freezing of gait and stuttering or stammering speech gradually develop. Axial rigidity with severe neck stiffness and the lack of limb rigidity are distinctive features. Eye movement abnormalities, blepharospasm, postural instability, and falls occur significantly later in PSP-PAGF than in RS. Supranuclear vertical gaze palsy is a late feature and may be entirely absent in some cases. Pronounced frontal subcortical impairment, bradyphrenia, asymmetric bradykinesia and rigidity, tremor, or levodopa response are not associated with the PSP-PAGF subtype.
The full clinical syndrome of PAGF is highly predictive of PSP–tau pathology. A study reported that among 759 patients with parkinsonism, 7 had a full clinical syndrome of PAGF, of whom 6 had PSP–tau pathology and 1 had Lewy body Parkinson’s disease (54). Other conditions, including subcortical white matter ischemia (Binswanger leukoaraiosis), normal pressure hydrocephalus, and dementia with Lewy bodies, may also present with isolated gait freezing (55). The prognosis of PSP-PAGF is more benign than RS, and the median disease duration is 11 years (3).
PROGRESSIVE SUPRANUCLEAR PALSY–CORTICOBASAL SYNDROME
CBS is the classic presentation of corticobasal degeneration (CBD), a condition that shares pathologic similarities with PSP. It is now apparent that only 25% to 56% of patients presenting with CBS have pathology consistent with CBD (56). A United Kingdom Brain Bank series showed that there were heterogeneous pathologic causes for the 21 cases who had received a final clinical diagnosis of CBS: 5 (24%) had CBD, 6 (29%) had PSP, 5 (24%) had Alzheimer’s disease, 3 (14%) had frontotemporal lobar degeneration (FTLD), and 2 (9%) had Parkinson’s disease (57). Overall, 70% of CBS cases have an underlying tauopathy. While PSP is one of the most common causes of CBS, CBS is a rare clinical presentation of PSP and the PSP-CBS subtype comprises of only 4% of all PSP cases (22,58).
Patients with CBS typically present with progressive clumsiness and loss of function of one hand due to a combination of frontoparietal and basal ganglia sensorimotor dysfunction. Ideomotor apraxia, parietal sensory dysfunction, focal limb dystonia, action- or stimulus-sensitive myoclonus, levodopa-unresponsive rigidity and bradykinesia, and alien limb phenomenon may all be found on examination and contribute to the asymmetric limb dysfunction. Delayed initiation of horizontal saccades, a characteristic feature of CBS, can be observed in PSP-CBS, and is more pronounced when gaze is directed toward the side of the apraxic limb. Progressive nonfluent aphasia (PNFA) and apraxia of speech (AOS) are common associated features (59). Pyramidal and Babinski’s signs are observed in half of the PSP-CBS cases (22).
The pronounced asymmetric clinical presentation in PSP-CBS is distinctive from RS, which is typically symmetric and axial predominant (22). Cortical signs are a prerequisite for the diagnosis of CBS, which should not be made based on marked asymmetric motor disability or focal limb dystonia alone without signs of cortical sensory loss or limb apraxia. Some PSP-CBS patients have a pure clinical picture of CBS, which makes antemortem prediction of underlying PSP pathology difficult. Occasionally in PSP-CBS, there are overlap of clinical features with RS. Postural instability in the first year of disease onset and supranuclear downgaze palsy in patients with CBS are clinical pointers of underlying PSP rather than CBD pathology, even though there are also signs of CBS. The prognosis of PSP-CBS is similar to RS, and the median disease duration is 7.3 years (22).
PROGRESSIVE SUPRANUCLEAR PALSY–PROGRESSIVE NONFLUENT APHASIA
PNFA is a language disorder characterized by nonfluent and effortful speech with hesitancy, agrammatism, and phonemic errors. AOS frequently occurs together with PNFA but can present in isolation (59). The primary features of AOS are slow, segmented and groping speech with errors in timing and coordination, abnormal prosody, and decreased phonetic accuracy. PNFA and AOS may occur in patients with otherwise-typical clinical syndrome of RS or CBS. When PNFA and AOS are the dominant presenting features, it is strongly predictive of an underlying PSP or CBD pathology (60,61), and is associated with the late emergence of other characteristic signs such as supranuclear vertical gaze palsy or limb apraxia (62). PNFA with less-prominent AOS is more likely to be due to CBD or FTLD-TAR DNA-binding protein-43 (FTLD-TDP). Progressive agraphia may sometimes precede the onset of these speech deficits (63). PSP-PNFA represents another PSP subtype characterized by asymmetric cortical presentation involving the dominant hemisphere and has similar age of onset and disease duration as RS (61).
PROGRESSIVE SUPRANUCLEAR PALSY–BEHAVIORAL VARIANT OF FRONTOTEMPORAL DEMTIA
Behavioral variant of frontotemporal dementia (FTDbv) is typically associated with FTLD-TDP but may also be caused by Pick’s disease, CBD, PSP, or Alzheimer’s disease (64,65). The PSP-FTDbv subtype is rare and represents 4.5% of all PSP cases in one study (66). Likewise, the clinical syndrome of FTDbv is more commonly associated with non-tau pathologies, and only less than 4% of cases with FTDbv had PSP pathology (67). Prominent insidious onset of behavioral and personality changes dominates the clinical presentation, and strikingly disproportionate behavioral changes can remain the most prominent feature throughout the disease course in PSP-FTDbv. Emotional blunting, aggressive outbursts, distractibility, hyperphagia, neglected hygiene, socially inappropriate, disinhibited, and compulsive behaviors, and the loss of insight contribute to significant distress to family members. Postural instability, falls, supranuclear vertical gaze palsy, micrographia, and dysphagia may emerge in the later stages, serving as clinical pointers to underlying PSP pathology (66).
DIFFERENTIAL DIAGNOSIS
When assessing patients with postural instability, falls, and eye movement abnormalities, other conditions should be considered (Table 14.4). Magnetic resonance imaging (MRI) of the brain may provide support for the diagnosis of PSP, with the identification of midbrain and/or superior cerebellar peduncle atrophy. It is also very important in helping to exclude extensive cerebrovascular disease (68,69), normal pressure hydrocephalus (55), FTLD or a structural midbrain lesion (70,71), which can all masquerade as PSP. Further tests, including syphilis (72) and HIV serology (73), autoimmune profile (74), and a screen for paraneoplastic disease (75), can be considered in individual cases to exclude potentially treatable conditions. Whipple’s disease is a rare but potential treatable condition that can present with a gaze palsy and may be diagnosed by small bowel biopsy or cerebrospinal fluid (CSF) polymerase chain reaction for Tropheryma whippeli (76). In patients with rapid disease progression, the findings of CSF 14-3-3 protein, cortical high signal on diffusion-weighted MRI images and diffuse slowing of electroencephalogram, or periodic triphasic waves are supportive of the clinical suspicion of sporadic Creutzfeldt–Jakob disease (77). CSF biomarkers for Alzheimer’s disease, including an elevated total tau protein and reduced β-amyloid 1-42 protein, may point to an underlying diagnosis of Alzheimer’s disease (78). An early disease onset, positive family history, or concurrent clinical syndromes (e.g., motor neuron disease or FTD) may point to a genetic phenocopy of PSP related to mutations in MAPT (79), progranulin (80) or C9orf72 repeat expansion (81).
| Differential Diagnosis of PSP and PSP Look-alikes |
Neurodegenerative disorders | Parkinson’s disease, multiple system atrophy–parkinsonism, corticobasal degeneration, sporadic Creutzfeldt–Jakob disease, dementia with Lewy bodies, chronic traumatic encephalopathy, postencephalitic parkinsonism |
Genetic or heredodegenerative disorders | MAPT, progranulin, C9orf72, Wilson’s disease, Niemann Pick type C, Kufor–Rakeb syndrome, adult-onset Gaucher’s disease, Perry’s syndrome, LRRK2, POLG, SCA2, SCA3, SCA7, Huntington’s disease–Westphal variant, DRPLA, neuroacanthocytosis |
Infections | Whipple’s disease, neurosyphilis, HIV |
Acquired or drug-induced disorders | Manganese and ephedrone intoxication, acquired hepatocerebral degeneration, hypoxia, neuroleptics, Wernicke’s encephalopathy |
Vascular and white matter abnormalities | Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, cerebrovascular disease, CSF1R |
Structural abnormalities | Midbrain tumor, normal pressure hydrocephalus |
Paraneoplastic causes | Paraneoplastic midbrain encephalitis (particularly associated with Ma2 antibodies) |
Autoimmune causes | Antiphospholipid syndrome, myasthenia gravis, neurosarcoidosis |
NEUROIMAGING
The typical MRI findings in PSP include atrophy of the midbrain and superior cerebellar peduncle and dilatation of the third ventricle (82). Quantitative measures of the diameters of midbrain and pons on midsagittal T1-weighted MRI may improve diagnostic accuracy. A study showed that in healthy controls, the mean midbrain ratio was approximately two-thirds of the pontine diameter, while in PSP it was <52%, and in MSA, the ratio was greater than two-thirds (83). A combination of midbrain diameter of <9.35mm and ratio of <0.52 had a 100% specificity for PSP and may reliably differentiate PSP from MSA. The “hummingbird sign” or “giant penguin sign” on midsagittal plane, with rostral midbrain atrophy representing the “beak,” may be seen in 67% of PSP cases (82,84) (Fig. 14.1). The “morning glory sign” on axial images, due to concavity of the lateral margin of the midbrain tegmentum, is another specific but less-sensitive feature (82,84) (Fig. 14.2). A study of diffusion-weighted MRI evaluating regional apparent diffusion coefficient (rADC) demonstrated an increased putaminal rADC reliably distinguished early PSP from Parkinson’s disease, with a sensitivity of 90% and a positive predictive value of 100%, but not MSA-P (85).
Functional imaging techniques, such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT), may be useful in supporting the diagnosis of PSP. PET imaging using the [F-18]FDDNP probe can identify fibrillar tau deposits in the living human brain. A study suggested that [F18]FDDNP binding in the midbrain and subthalamic regions distinguished PSP from Parkinson’s disease and healthy controls, indicating its potential as a radiologic biomarker in the follow-up of disease progression in future tau-specific therapeutic trials (86). Dopamine transporter SPECT imaging shows severe striatal reduction in PSP and may be a useful tool to differentiate PSP from some of the PSP look-alikes, such as cerebrovascular disease and normal pressure hydrocephalus (87) (Fig. 14.3).
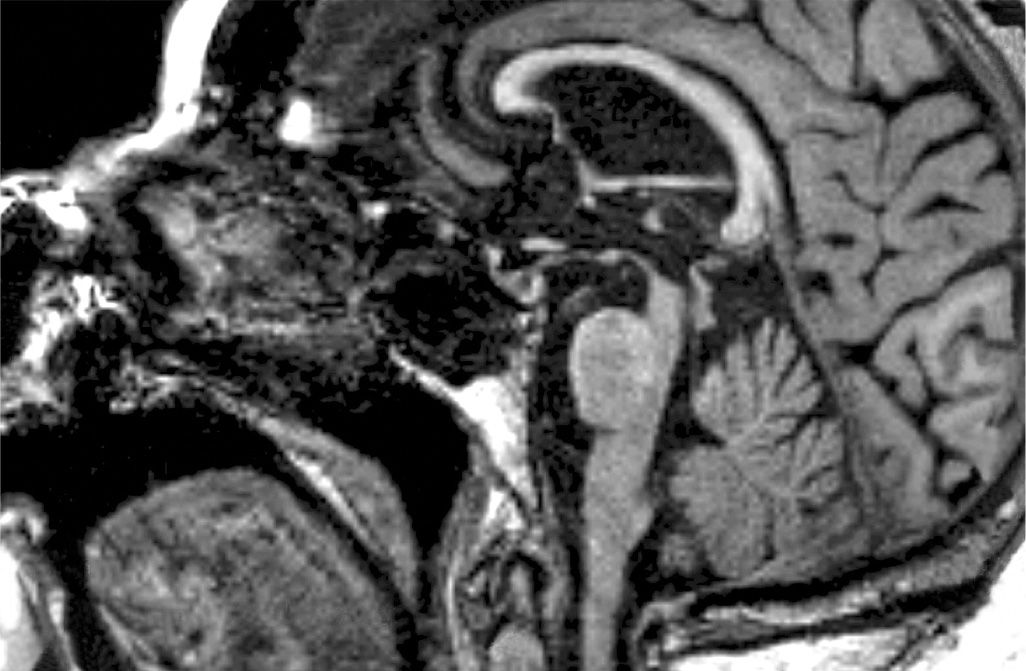
Figure 14.1. The “hummingbird sign” in PSP. Midsagittal T1-weighted MRI appearances of the brain stem, showing a concave superior border of the midbrain that is disproportionately atrophied compared with the pons. Striking hyperextension of the neck is another characteristic feature on sagittal MRI.








