(1)
Asheville, NC, USA
The reversible folding and unfolding of proteins uncovered by Anfinsen and Pauling was a landmark event. It established the primacy of the amino acid sequence in determining the functional, native state of a protein. It further set the stage for the equally remarkable discovery that proteins can possess more than one stable conformation; and that these alternative conformations, brought on by mutations or abnormal environmental conditions, can promote aggregation and a host of neurodegenerative diseases.
This second fundamental idea took over 30 years to emerge. Several key discoveries led the way. Chief among these were the 1959 electron microscope experiments of Cohen and Calkins followed by the 1968 X-ray diffraction studies by Glenner and Wong. These experiments established that amyloids were composed of sets of β-sheets oriented parallel to the amyloid fibril axis with each strand in each β-sheet oriented perpendicular to the fibril axis thereby producing a characteristic cross-β pattern.
Additional studies repeated over several years with different proteins established that many if not most proteins had the capability to adopt this alternative form if their environment was suitably manipulated. Remarkably, the proteins that underwent this change in conformation had nothing in common. These changes occurred whether the proteins were large or small, globular or extended, predominately beta-sheet or alpha-helical. The conclusion drawn from these studies was that the ability to adopt the cross-β-generating conformation was a basic property of the protein backbone. Under normal conditions the adoption of an alternative, potential-disease-causing conformation does not occur. In stark contrast, in the neurodegenerative disorders, one or more proteins fail to properly fold and remain in their native state and instead adopt the alternative cross-β conformation and collect in insoluble extracellular and intracellular deposits.
A major challenge in the study of neurodegenerative disorders has been, and remains, to firmly establish the causal chain linking the observed amyloids and other kinds of deposits of misfolded proteins to the disease severity and progression. In the case of the systemic amyloidose s discussed in Chap. 1, the broad features if not the details are becoming clear. The misfolded proteins form intrusive deposits in cells, tissues, and organs leading to their malfunction, failure, and death. Not so in the case of Alzheimer’s disease or Parkinson’s disease or any of the other neurodegenerative disorders. The correlations between the visible deposits (Table 4.1) and disease are far more complex. They are made so by the presence of multiple types of aggregates that can and do interconvert into one another. A minimal list of the types of structures formed by misfolded proteins is, as follows:
Table 4.1
Prominent disease-causing proteins and the visible deposits they form
Protein | Deposit | Site of action | Disease |
|---|---|---|---|
Aβ | Amyloid plaques | Temporoparietal | AD |
α-Synuclein | Lewy bodies Lewy bodies | Midbrain Frontotemporal | PD LBD |
tau | Tangles Pick bodies | Temporoparietal Frontotemporal | AD FTLD |
Huntingtin | NIIs | Basal ganglia | Huntington’s |
SOD1 | Bunina bodies | Motor cortex | ALS |
TDP-43, FUS | Inclusion bodies Inclusion bodies | Motor cortex Frontotemporal | ALS FTLD |
Monomers
Soluble oligomers
Amorphous aggregates
Amyloid fibrils
Monomers, single molecules that alter their conformation from that of the native state, are the natural starting point in development of a protein misfolding disorder . These may append to a growing fibril or alternatively aggregate into small, soluble oligomeric complexes containing two, three, four, or more misfolded proteins that may or may not lie on the pathway leading to amyloid fibrils . There is emerging evidence that in many instances these small assemblies may be more toxic than the large insoluble deposits found in inclusion bodies and extracellular spaces. In addition, not all misfolded proteins generate amyloids. Misfolded huntingtin and TDP-43 do not do so. Instead, they accumulate in inclusion bodies as amorphous aggregates with varying degrees of amyloid-like properties.
A major difficulty in looking deeper into the structure of the amyloids was their large insoluble character. A way around this difficulty presented itself through the realization that the core structure of the amyloids might be generated from short fibril-generating sequences of just six or seven or eight amino acid residues. These findings led in 2005 to the first atomic level three-dimensional amyloid fibril crystal structures built from these short sequences. This chapter will begin with an examination of amyloid microcrystals and with the two leading models of how fibrils they might naturally develop.
There are numerous as yet unanswered questions on the road to effective therapeutics. One paramount set of questions has as its focus the identity of the toxic species responsible for each of the neurodegenerative diseases, and how these species might be produced. A key concept is of their development through loss of stability and partial unfolding; another is the contributing role of protein fluctuations and perturbations. An examination of oligomer formation by these means will follow the exploration of fibril structure and formation.
Many proteins especially those involved in cell regulation and signaling, the sine qua non of neurons, contain large unstructured regions and remain unfolded in their native state. These polypeptides are referred to as intrinsically unstructured proteins (IUPs) or even more commonly as intrinsically disordered protein s (IDPs). Numbered among this group are many of the proteins with prominent roles in neurodegeneration, including most, if not all, of the entries in Table 4.1. These proteins contain large regions lacking any discernable secondary structure. This chapter will conclude with a first look at this class of proteins.
4.1 Structure of the Amyloid Fibril
The first microcrystal structure solved in 2005 was that of the yeast protein Sup35. This structure is presented in Fig. 4.1. In this figure, one observes the cross-β spine of the amyloid fibrils formed by the seven-residue sequence GNNQQNY that is rich in glutamine (Q/Gln) and asparagine (N/Asn) amino acids. Repeating segments of this sequence assembled within several hours in solution into the pair of parallel beta-sheets shown in Fig. 4.1a. Each sheet was erected from identical seven-residue strands exactly in register, stacked one on top of another.
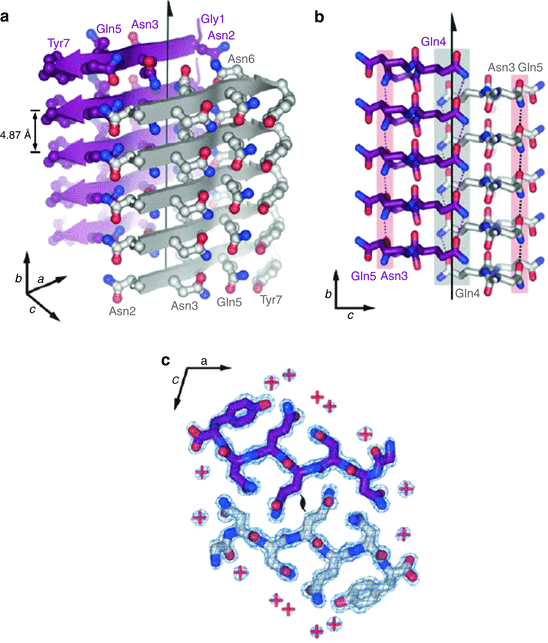
Fig. 4.1
Amyloid fibrils formed by the seven-residue sequence GNNQQNY. (a) Stacking of the monomers one on top of the other along the fibril axis (indicated by the vertical arrow). (b) The two parallel sheets are shifted relative to one another to permit the interdigitization of the side chains. (c) Top-down view showing the locations of the water molecule indicated by red plus signs showing that water molecules are absent from the interface (from Nelson Nature 435: 773 © 2005 Reprinted by permission from Macmillan Publishers Ltd)
An extensive network of hydrogen bonds is formed within each beta-sheet. Some of these hydrogen bonds are between backbone residues, while the others are amide-amide hydrogen bonds formed between identical pairs of polar Asn and Gln residues in adjacent molecules. Recall that glutamine and asparagine side chains end with a hydrogen-acceptor oxygen atom and a hydrogen-donor amide group. These side chains enable the formation of “amide stacks” within each sheet. This arrangement of parallel beta-sheet-forming residues was given the name “polar zipper” by Max Perutz in a 1993 study of how long polyglutamine repeats prevalent in Huntington’s disease and other polyQ disorder s might form parallel beta-sheet-like structures.
The second important feature of this structure is that the two beta-sheets are held together not by hydrogen bonds but instead by van der Waals forces between polar side chains on beta-strands belonging to adjacent β-sheets. These side chains are interdigitized with one another (Fig. 4.1b). This arrangement was given the name “steric zipper” in analogy to Perutz’s polar zipper. The third key feature of the structure is that water molecules although readily present elsewhere are expelled from the interface, which is tight and dry (Fig. 4.1c).
One of the most significant set of findings preceding the atomic level structures were the apparent universality of the amyloid fibril structure. A large number of polypeptides, irrespective of their primary sequences, could be made to refold into amyloid-forming conformations. Some of the proteins were small; others were large. Some folded normally into tight globular shapes while others assumed more extended forms. One conclusion drawn from these observations is that the ability of these proteins, or portions thereof, to fold into amyloid-templating structures is a general property of the polypeptide backbone. The sequence dependence, which under normal circumstances would play a key role in determining the fold, still plays a role albeit a different one. Its role in this alternative folding universe manifests itself at the atomic level through the existence of more than one kind of microcrystalline structure.
This point was brought out in studies that examined the spines of amyloid fibrils generated by different short peptide sequences. It was found, for example, that eight different kinds of steric zippers can be generated by short peptide amyloidogenic sequences. Examples studied included sequences from Aβ, tau, Sup35, and insulin. The eight (2 × 2 × 2) different classes are defined by (1) orientation of their faces (face-to-face or face-to-back); (2) orientation of their strands (up-up or up-down); and (3) whether the strands within each sheet were oriented in parallel or antiparallel.
Further variability in the fibril structures arises from their dependence upon their surroundings. The same sequences can assemble into several different microcrystalline forms as the growth and environmental conditions are altered. This dependence may include influences of the full chains. There is evidence that the microcrystalline forms represent reasonably well the actual in vivo fibrillar structures, but this remains to be established firmly. What can be said is that without question the forms generated by amyloid-competent sequences are highly polymorphic and responsive to their surroundings.
A second 2005 amyloid fibril atomic structure was that of the amyloid-β peptide generated from H/D-exchange NMR. It is presented in Fig. 4.2. Whereas the sequence shown in Fig. 4.1 was rich in polar residues, the amyloid-β fibril is dominated by hydrophobic residues. The Aβ fibrils have as their core two dissimilar sequences connected by a flexible loop resulting in U-turn heterozipper. The fibril-generating core consists of the hydrophobic segment containing residues 18–26 that comprise the β1 strand and residues 31–42 that form the β2 strand.
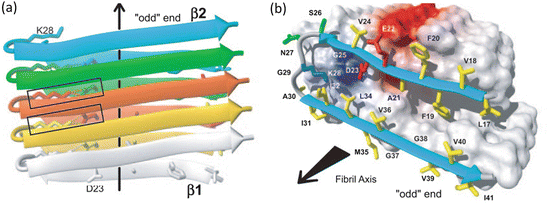
Fig. 4.2
Amyloid fibrils formed by residues 18–26 and 31–42 from Aβ (1–42) as determined by means of hydrogen-deuterium exchange and NMR. Arrows denote the fibril axis. (a) Parallel, in-register stacked strands form two β-sheets along the fibril axis indicated by the arrow. (b) Hydrophobic, polar, negatively charged and positively charged amino acid side chains are colored yellow, green, red, and blue, respectively; all others are depicted as white (from Lührs PNAS 102: 17342 © 2005 National Academy of Sciences, U.S.A. and reprinted with their permission)
Yet another atomic level three-dimensional fibril structure is shown in Fig. 4.3. This structure, determined by means of solid state NMR, was of the prion protein HET-s (218–289). In this case, the β-sheets form a solenoid-like structure in which each monomer contributes two windings. The structure possesses a compact triangular core of hydrophobic residues along with two asparagine ladders, several salt bridges, and a large number of hydrogen bonds. The core contains three strands per winding resulting in parallel in-register β-sheets aligned along the fibril axis. The overall structure is a highly stable one and far less subject to polymorphic variability than many of the other structures.
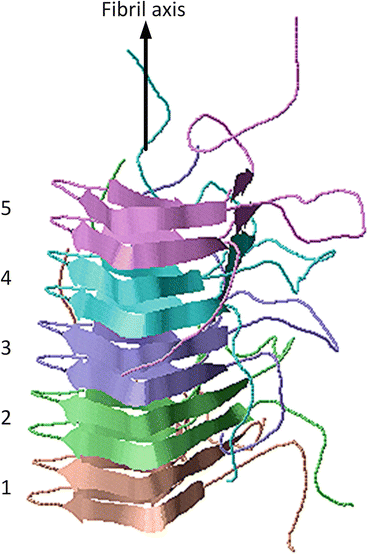
Fig. 4.3
Amyloid fibrils from the fungal prion protein HET-s (218–289), which forms a triangular-shaped β-solenoid. The grey arrow denotes the fibril axis. The five winding pairs are color coded. The figure was prepared using Jmol with atomic coordinates deposited in the PDB under accession code 2NMR
4.2 Amyloids as Biomaterials
William Astbury coined the term “molecular biology” in 1950, in his Harvey Lectures, to describe the new emerging field concerned with the forms of biological materials, that is, with their three-dimensional structure and their genesis and function. This field had as its cornerstone the X-ray crystallography studies of the Bragg’s, of Bernal and Pauling and Kendrew and Perutz, along with Astbury, and all their essential collaborators and colleagues too numerous to mention. Now, some 80 years after Astbury’s examinations of keratin, amyloids in their various aggregated forms are seen as the causes of neurodegenerative diseases, and another new field that of engineered biomaterials for tissue and organ repair is in its early stages of development.
Spider silk fibrils are remarkable structures. They, like amyloids, are assembled from beta-strands oriented perpendicular to the fibril axis and held together by hydrogen bonds. Although technically not amyloids they are similar, both belonging to a large class of structures erected from simple polypeptide building blocks. Like amyloids the spider silks are held together by cooperative networks of hydrogen bonds. Although these bonds are individually weak far more so than covalent bonds the overall effect of hydrogen bonding is to generate materials that are remarkably strong. In addition, whereas covalent bonds once broken stay broken, hydrogen bonds that break can reestablish themselves and are thus capable of self-repair. However, there are geometric/size limits to the stiffness and stability of hydrogen bonded amyloids and spider silk. Once individual strands and fibrils exceed a critical length they become weaker, more brittle, and less resilient to mechanical stresses. Fragmentation can then occur; this secondary process has important consequences not just for nucleating additional fibril growth but also for prion-phenomena as will be discussed later.
Amyloids, dragline silk, and other hydrogen bonded materials have high bending rigidity. That point is illustrated in Fig. 4.4 in which bending rigidity is plotted against the moment of inertia for a wide variety of materials. Recall that Young’s modulus, Y, also known as the elastic modulus, E, is a material-specific measure of stiffness. It is related to bending rigidity, B, and the (area-specific, cross-sectional) moment of inertia, I, through the expression B = Y ∙ I. The data points shown in the figure represent values for the Young’s modulus. These fall in several bands, according to the nature of the intermolecular forces holding the fibers together. Data points for a variety of amyloidogenic proteins are found in the orange band; their values are displayed in Fig. 4.4 as blue dots with error bars.
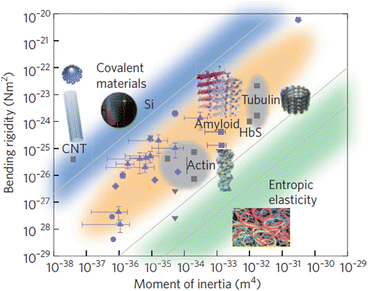
Fig. 4.4
Plot of bending rigidity versus the moment of inertia for different classes of materials. The blue band shows the range of values for metallic and covalently bonded substances; the orange band encompasses representative values for strong non-covalently bonded (e.g., hydrogen-bonded) structures. The green band contains values of weak non-covalently bonded materials. Blue symbols represent amyloidogenic substances; other types of materials are depicted in grey (from Knowles Nat. Nanotechnol. 6: 469 © 2011 Reprinted by permission from Macmillan Publishers Ltd)
4.3 Amyloids in Normal Physiology
Given their remarkable physical properties it is perhaps not surprising that cross-β amyloids fibrils have a role in normal physiology. They are utilized across multiple kingdoms—in bacteria, in yeasts, and in humans. In bacteria, they are secreted and function as an essential component of biofilms. Bacteria such as Escherichia coli (E. coli) are highly adaptable and can reversibly switch between a free-swimming planktonic lifestyle and a surface-attached, communal existence. Under stressful conditions they switch their gene expression programs to support construction and participation in communal single or multispecies biofilms. The biofilms contain not only the bacteria but also a protective, extracellular matrix of secreted molecules that facilitate attachment to surfaces and provides protection against agents in the environment potentially harmful to the bacteria. In many instances, amyloidogenic fibers are a major component of the biofilm extracellular matrix. Like the amyloids in neurodegenerative diseases, these fibers bind Congo red, exhibit birefringence, and possess an underlying cross-beta-sheet organization.
Bacterial amyloids are not a result of protein misfolding but instead are produced through a highly regulated assembly process, and, when desired, can be systematically disassembled to enable the bacteria to resume a planktonic lifestyle. The E. coli amyloidogenic materials are referred to as Curli fibers. Two Curli subunits—CsgA and CsgB—are centrally involved. The fibers themselves are composed of repeating CsgA units while the CsgB subunit directs their surface attachment, nucleation, and polymerization. The CsgA subunits contain an amyloid core domain consisting of a series of repeats enriched in glutamine and asparagine residues that generate the cross-beta-structure. These residues create a hydrogen bonded network that provides stability to the assemblage as they do for Sup35 discussed earlier in this chapter.
Functional amyloid s are also encountered in yeasts where they facilitate adhesion to solid surfaces, cells, and the extracellular matrix, and promote formation of biofilms. One particularly well-studied example is that of the human opportunistic fungal pathogen Candida albicans. In these studies, atomic force microscopy was used to explore how the cell surface Als adhesins mediate fungal attachment. A key finding is the presence of force-activated partial unfolding that exposes seven-residue amyloidogenic sequence IVIVATT. This sequence is highly enriched in hydrophobic (I, V) and Cβ-branched amino acids (I, V, T) that because of their particular side chains are ill-disposed to form alpha-helices and instead strongly favor formation of beta-strands. The beta-strands are stacked in amyloid fashion on top of one another to supply strong adhesive forces along with a stretching ability.
A question that naturally arises is how are the toxic effects of the functional amyloid s being avoided? One answer is that in humans the amyloids are sequestered in membrane-bound compartments thereby limiting their potential toxic effects. One example of this is the storage of peptide hormones in secretory granules, another is the melanosome matrix protein (Pmel17) fibers critical for melanosome maturation. In the former instance, endocrine hormones form stable amyloids while they are stored in secretory granules. Once they are released the large structures rapidly dissociate into monomers and are secreted from the cell. Thus, the potentially toxic amyloids are not only being encapsulated and sequestered, but they also readily dissociate unlike, for example, the Aβ peptides . In the second example, the Pmel17 amyloid fibers are also packaged in membrane-bound organelles, and in this case the melanosomes are subject to fast kinetics, both aspects limiting any toxic effects from oligomers and other potentially harmful intermediate sized aggregates.
4.4 Amyloid Growth Through Nucleated Polymerization
How repeating structures such as three-dimensional metallic crystals and one-dimensional amyloid fibrils self-assemble has been the subject of study for a century or more. Two models, or pictures, of how these structures develop have been put forth over the years. The older of the two is the nucleated polymerization picture inspired by how crystal growth occurs and how actin or tubulin fibers develop. This model was proposed as the basis for Aβ and prion amyloid growth by Jarrett and Lansbury in 1992. The other, nucleated conformational conversion , was put forth by Serio in 2000; it was suggested by the presence of significant numbers of structurally fluid prion oligomers.
In crystal growth, atoms undergo a phase change from a three-dimensional disordered ensemble to a highly ordered spatially regular arrangement. This process takes place in several stages. In the, initial, slow stage a seed had to form that serves as the nucleus for the subsequent and far more rapid crystal growth stage. In a series of papers published in the 1939–1941 time period, Melvin Avrami derived a simple expression, the Avrami equation, for the crystallization kinetics:
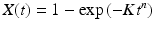
In this expression, X(t) is the fraction of atoms that have transitioned to the crystalline phase at the given time t, K is a geometry-dependent growth rate, and n is an integer whose value varies with the type of nucleation and structure of the crystal. Similar expressions to this one have been applied to the growth of amyloid fibrils .
(4.1)
Sickle cell anemia is a disorder brought on by a single point mutation in the gene coding for the beta-chain of hemoglobin. This mutation results in the substitution of valine for glutamic acid at position 6; this alteration has no effect on the fully oxygenated state of hemoglobin, but results in a reduced solubility of deoxygenated hemoglobin, HbS. As a result HbS forms aggregates referred to as a gel that deforms the normal red blood cells into a crescent or sickle shape and results in vascular occlusion and decreased blood flow through the microcirculation. Under an electron microscope the fibrils comprising the gel are found to be organized into two helical layers—an inner layer of four polymeric strands surrounded by a second helical layer consisting of ten strands. The result is a 21 nm diameter structure consisting of 14 strands twisted together to form long fibrils.
Sickle cell disease was first described in 1910 by the physician James Bryan Herrick (1861–1954) who had noted the odd-shaped cells. Further studies by Herrick and others over the next 15 years or so further established the connection between the sickle-shaped red blood cells and the anemia. That set the stage in 1949 for the landmark paper by Linus Pauling that identified an aberrant form of hemoglobin as the causal agent of the disease and introduced the concept of “molecular medicine”, and the study by James Neel (1915–2000) that established its genetic origins.
Another set of early pioneering studies focused on normal growth in the cell of tubulin and actin filaments. Those considerations led to the adaptation of crystal growth ideas into the realm of one-dimensional protein polymerization by the Japanese physicists Fumio Oozawa and Sho Asakura. Their investigation was followed a short time later by a series of studies by William A. Eaton and coworkers in the 1970s and 1980s on HbS polymerization and the further expansion of Oozawa and Asakura’s model of nucleated polymerization to include secondary polymerization. The overall joint result is a central model of how both normal and abnormal filaments and amyloids develop. The key mechanisms underlying protein nucleated polymerization are illustrated in Fig. 4.5.
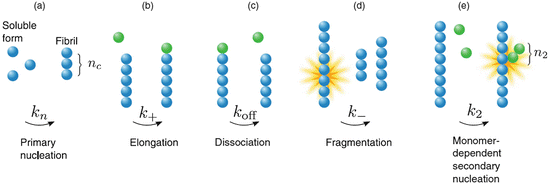
Fig. 4.5
Nucleation-polymerization mechanisms. (a) Primary polymerization leading to formation of a filament of length n c; (b) linear growth from either end of the elongating filament; (c) loss of monomers from either end of the filament; (d) fragmentation , and (e) secondary nucleation leading to formation of branch (secondary) filament of length n 2 (from Cohen 2011 J. Chem. Phys. 135: 065106 © 2011 AIP Publishing, LLC and reprinted with their permission)
This model describes several features of significance. First, the initial stage, referred to as a lag phase, is a slow one since a number of nuclei or seeds have to form first, and this is kinetically unfavorable. Once nuclei have formed the process speeds up with the addition of monomers to either end of the growing one-dimensional fiber. As noted in the section on amyloids as a biomaterial , once the fiber length exceeds a critical value it becomes increasingly brittle and prone to shed pieces. Several studies have been carried out that establish that fragmentation and secondary nucleation are important contributors to amyloid pathology. Lastly, in the nucleated polymerization model, the concentration of amyloid-competent monomers must exceed a threshold; there are few oligomers smaller than a nucleus, and the lag phase can be shortened and perhaps eliminated if seeds are externally supplied at the outset.
4.5 Amyloid Growth through Nucleated Conformational Conversion
Oligomers are non-covalently-attached assembles of several identical monomers. These small aggregates are quire varied in their numbers and in their geometric arrangements (morphology). Some form pore-like water-bearing rings, others are cylindrical in shape but have a dry interior. Pathway destinations are of considerable importance. Some oligomers are amyloidogenic and serve as aggregation intermediates on the pathway towards amyloid assembly. Others are, in themselves, aggregation endpoints and may be toxic or not.
Oligomers are the main components in an alternative model of how amyloid fibrils may develop. In this second model oligomers serve as the locus of the conversion from non-amyloid-competent to amyloid-competent units. This process is one in which monomers initially and rapidly associate into amorphous oligomers. The units within these aggregates then undergo a slow nucleated or templated conformational conversion through their interactions with one another into ordered beta-strands and sheets that self-assemble into the fibrils as depicted in Fig. 4.6.
Fig. 4.6
Nucleated conformational conversion . Blue bent monomers aggregate into oligomers which then serve as a “reactor” for the conversion of the oligomers into beta strands depicted as yellow arrows (from Blieholder Nat. Chem. 3: 172 © 2011 Reprinted by permission from Macmillan Publishers Ltd)
This model has a different kinetics from that of the nucleated polymerization model and a different dependence on concentration and associated thresholds. For example, unlike the nucleated polymerization model a significant number of soluble oligomers are present early in the fibril-forming process. Several recent studies of amyloid formation have been carried out that support this more recent oligomer-centric model emphasizing as they do the appearance of significant numbers of oligomers before the first fibrils appear.
4.6 The Toxic Oligomer Hypothesis
The “amyloid cascade hypothesis ” appeared in a mature form in a 1992 paper by Hardy and Higgins. This hypothesis posits that deposits of Aβ peptide in the brain are the crucial events leading to Alzheimer’s disease. This has been the basic operating principle behind much of the activity in the field during that past 20-plus years. However, identification of the specific form or forms of Aβ responsible for causing dementia is not clear. The primary object of interest has been the extracellular amyloid plaque s , but clinical correlations between the extracellular deposits and the disease progression are not convincing and, in many cases, are absent. The presence of tau and the tangles added another level of complication as does the natural conversion of one form to the other through growth and fragmentation .
In an update and revision to the amyloid cascade hypothesis , toxic oligomer s have been hypothesized to be the primary causative agents in Alzheimer’s disease and other forms of neurodegeneration. These smaller soluble species are far more mobile and expose a greater fraction of hydrophobic surfaces than do the large insoluble amyloids and amorphous aggregates. Support for this shift in attention is provided by investigation of several different diseases. One of these is type 2 diabetes. In this disorder, islet amyloid polypeptide (IAPP) accumulates in the pancreas. These deposits were initially believed to be the main cause of β-cell failure paralleling the finding for most if not all the systemic amyloidosis. But difficulties arose with ascribing the cause of the disease to the extracellular deposits. The main event in the disease progression is death of β-cells through apoptosis. However, that this was due to the extracellular deposits of IAPP seemed to be implausible. Evidence that oligomers might be capable of permeating the cell membrane thereby triggering a series of events leading to apoptosis soon followed. The idea that oligomers might be the toxic agent in type 2 diabetes was further strengthened by the lack of a similar effect on membranes by components of the extracellular IAPP deposits.
A series of reports that appeared in the decade from 2002 to 2011 provided further evidence for a major role of toxic oligomer s in Alzheimer’s disease and other neurological disorders. Two biophysical features of these oligomers—their expose of hydrophobic surfaces and the presence of regions of intrinsic disorder in the candidate toxic proteins and their interaction partners—were centrally implicated in their ability to cause harm to and kill cells. Complicating the issue of the identity of specific toxic oligomers is their tremendous variability in oligomer size, shape, density, and hydrophobicity. Heterogeneous mixtures of monomers, dimers, trimers, tetramers, and so on up to and including large amorphous aggregates and fibrillary deposits can occur. The fractional contribution of each type of small oligomer and large aggregate to a given mixture at any time depends critically on the particular species and its biophysical properties.
The identification of certain features of the oligomers as being important was made possible by the creation of agents that recognize specific-binding epitopes on those proteins. Among these are antibodies called OC and A-11, and the fluorescence probe 8-anilino-1-naphthalenesulfonic acid (ANS). As noted above, a variety of oligomeric assembles are encountered; these small soluble oligomers vary in size and morphology. Some of them, referred to as prefibrillar oligomers, bind A-11 while at the same time are negative to binding the fibril-specific antibody OC. Others, termed fibrillar oligomers, bind OC but not A-11. Interestingly, the A-11 antibody can select out oligomers of diverse sequence structure apparently recognizing a common epitope. Similarly, ANS, which is used to detect conformational changes, recognizes a common hydrophobic epitope in proteins possessing unrelated primary sequences. Furthermore, the strength of the binding of ANS to these proteins correlates well with their increasing toxicity.
There is yet another layer of complexity beyond that of the differing sizes and morphologies of the aggregates. That layer is associated with variations in the primary sequence from which oligomers and fibrils are generated. These differences arise in several ways: First, there are variations in primary sequence arising from alternative splicing. Secondly, cleavage of the full-length protein often occurs resulting in the generation of protein fragments. Thirdly, and obviously, mutations alter the primary sequence. Leaving aside for the moment the mutations, the following are examples of alterations in primary sequence associated with the aggregation-prone proteins listed in Table 4.1:
Aβ: Peptides vary in length from 39 to 43 amino acids; in addition, shortened peptides are produced in which a number of the N-terminal-most residues have been removed.
tau: Six isoforms exist resulting in proteins ranging in length from 352 to 441 amino acids; shortened proteins are formed through truncation of N- and C-terminal residues.
huntingtin: PolyQ tract lengths vary; full-length and N-terminal fragments are produced.
TDP-43: C-terminal fragments are generated.
These changes in primary sequence can profoundly alter the tendency to misfold, aggregate, and cause disease. Even small differences can have large effects. Shown in Fig. 4.7 are the results of a study of Aβ1–40 and Aβ1–42 oligomer formation. Of the two forms, Aβ1–42 is regarded as being far more toxic. As the figure shows there are differences in the sizes and morphologies of the corresponding oligomeric species formed, and these distinctions are thought to be linked to their varying disease-causing propensities.
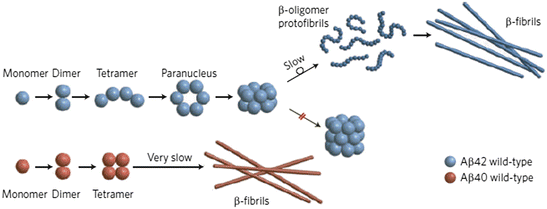
Fig. 4.7
Aβ1–40 (Aβ40) versus Aβ1–42 (Aβ42) oligomer and fibril formation (from Clemmer Nat. Chem. 1: 257/Bernstein Nat. Chem. 1: 326 © 2009 Reprinted by permission from Macmillan Publishers Ltd)
In many situations, the conformational variability of the putative toxic proteins creates challenges in identifying the exact toxic species responsible for a particular disorder. Those protein containing extensive disordered regions are particularly susceptible to these complications. This class of proteins, referred to as intrinsically disordered protein s , is heavily implicated as causative agents in neurodegeneration. Prominent examples include alpha-synuclein, tau, and huntingtin. Portions of SOD1, TDP-43, and FUS implicated in the frontotemporal dementia s and ALS are unstructured, as well. As notable example of this flexibility is α-synuclein, which justifies adding another entry to the above-mentioned bullet-list:
α-Synuclein: Called a chameleon because of its conformational diversity, this protein exhibits varying amounts of α-helical and β-sheet secondary structure.
Lastly, the relationship between oligomers and fibrils is a complex one that depends on the biophysical and biochemical details of the involved species. As seen in studies of Aβ aggregation, amyloid fibrils are variable in structure and may be generated from monomers and other small oligomers in more than one way. An added dimension to their complex interrelations is the existence of fragmentation and secondary nucleation pathways, processes, illustrated schematically in Figs. 4.5 and 4.8 that link together oligomers and fibrils. An important recent observation in this regard is that new oligomers may be generated, when both monomers and fibrils are present in sufficient numbers, by means of fibril surface-catalyzed (secondary) nucleation. For all of these reasons fibrils may yet play an important role in the pathogenesis of Alzheimer’s disease and the other amyloidogenic disorders.
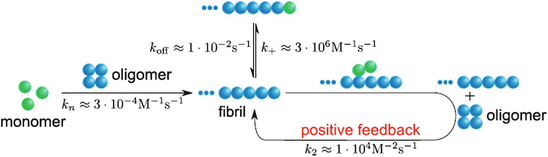
Fig. 4.8
Role of secondary nucleation in Aβ aggregation (from Cohen PNAS 110: 9758 © 2013 Reprinted with permission from Tuomas Knowles)
4.7 Coding Strategies that Prevent Misfolding and Aggregation
Physics and genetics work together to ensure that proteins that are expressed can fold into their native conformations in physiologically useful timeframes. As discussed in Chap. 2, they do not carry out exhaustive and time-consuming conformational searches; instead, they utilize a number of highly efficient macromolecular-force-driven folding stratagems. The interplay between physics and genetics goes much further. Sequences that are especially aggregation-prone are normally prevented from doing so by a variety of coding strategies. First and foremost, aggregation-prone portions of the protein tend to be buried in the core of the natively folded protein and only upon destabilization become exposed.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


