(1)
Asheville, NC, USA
The 1930s, 1940s, and 1950s were a time of change for how proteins were viewed. Results from X-ray crystallography and denaturation experiments guided the way from the idea that proteins were the repository of genetic information, and DNA provided structural support, to one in which DNA was the data repository. In the revised modern picture, proteins provided structure support and were the workhorses of the cell, performing most of the necessary cellular tasks.
Notions on the permanence of proteins also underwent major changes during this period. At the outset, it was believed that there were two kinds of proteins—those that were derived from foodstuffs and were degraded to provide fuel for the body and proteins that were used for structural and functional roles in the body. The latter were essentially permanent and required replacement only when damaged. The view of the permanence of proteins was widely accepted awaiting the advent of tools that could be used to follow them in space and time. The needed tool was provided by the discovery of heavy isotopes of stable elements by Harold C. Urey (1893–1981). As a result Rudolf Schoenheimer (1898–1941) and David Rittenberg (1906–1970) were able to use 15N radiolabeled amino acids to demonstrate that structural and functional proteins underwent cycles of synthesis and destruction.
The striking notion of dynamic protein turnover gained traction with the discovery of a compartment where cellular components could be safely degraded. The lysosome was uncovered by Christian de Nuve in the 1953–1955 time-period. Further studies over the next several decades established the full extent of the lysosomal/vacuolar system for transporting and degrading exogenous and endogenous proteins and organelles. However, it was also found that not all proteins were degraded in lysosomes. Instead, many if not most cellular proteins were degraded somewhere else. Furthermore, it was found that this other, nonlysosomal degradation system required metabolic energy (that is, it was ATP-dependent).
The long-sought-for system was the ubiquitin-proteasome system. It took another 20 years or so for that to be established. The road to those discoveries led through studies of how abnormal hemoglobin was degraded in reticulocytes, newly produced red blood cells that, importantly, do not contain lysosomes. These findings were reinforced by the discoveries on how unwanted proteins were degraded in bacteria. It was found that the nonlysosomal degradation system required ATP and involved conjugation of the small protein ubiquitin to the target substrate leading to degradation by the as yet unknown protease. Those discoveries were followed by the identification of the unknown protease as a large multisubunit molecular machine —the 20S proteasome in 1980 followed by the discovery of the 26S proteasome shortly thereafter. Those studies culminated with the award of the 2004 Nobel Prize in chemistry to the main discoverers of the ubiquitin-proteasome system (UPS), Israeli biochemists Avram Hershko and Aaron Ciechanover, and American biophysicist Irwin Rose.
The existence of protein turnover and the lysosomal and proteasomal degradation systems was just one or several major developments following the discovery of the cellular roles of proteins, RNA, and DNA. A second major development was the emergence of a molecular chaperone system that assisted in protein folding, unfolding, disaggregation, and oligomeric assembly. These nanomolecular machines are active in bacteria, yeasts, plants, and animals where they play essential roles in maintaining protein homeostasis, or proteostasis .
The unveiling of the molecular chaperones began in the early 1960s in Pavia where Ferruccio Ritossa was exploring the effects of a brief exposure to elevated temperature on Drosophila gene expression. What he found was that the elevated temperatures triggered a change in the overall gene expression pattern—some genes were expressed more strongly while others had their expression tuned down. That finding was followed in 1974 by the study by Tissières, Mitchell, and Tracy in which Drosophila subjected to heat shock triggered a pronounced increase in RNA transcription and protein synthesis of a particular set of polypeptides logically termed heat-shock proteins (hsps). The most pronounced of these increases was of a protein of molecular weight of approximately 70,000 Da and thereafter called Hsp70.
The uncovering of Hsp70 was followed a few years later, in 1978, with a report by Ron Laskey that hsps were required in the assembly of histones into nucleosomes. This finding went against the prevailing belief that subunit assembly was an autonomous process. In addition, Laskey had discovered that while the hsp nucleoplasmin assisted in assembling the nucleosomes it was not part of the final structure. For that reason he coined the term “chaperone” to describe this unexpected role.
Those findings were followed a few years later by a convergence of two independent lines of investigation on the existence of a second major family of heat-shock proteins—the chaperonins. One line of research centered on the assembly of the chloroplast protein Rubisco; the other involved the replication of bacteriophages within Escherichia coli. In brief, John Ellis had discovered the existence of a Rubisco large subunit binding protein that assisted in assembly of newly synthesized Rubisco subunits. Another protein, named GroEL was identified and studied by several groups. This protein, needed for phage-λ morphogenesis in E. coli, consisted of 14 subunits organized into two stacked heptameric rings forming a cage that encapsulated its substrate. It was accompanied by a co-chaperone, named GroES in bacteria (Hsp10 in eukaryotes). Once GroEL was sequenced and compared to the chloroplast protein the similarity between them led to establishment of the groEL/Hsp60 chaperonin family essential for the folding of newly synthesized proteins and their assembly into oligomeric complexes.
By the early 1980s the Hsp60 and Hsp70 families of heat-shock proteins had been uncovered in yeasts, plants, and man, the bacterial counterpart to Hsp70, called dnaK, having been found by the late 1970s. Their investigation had established that heat-shock proteins/molecular chaperones were (1) highly conserved across multiple kingdoms; (2) were essential, and (3) were induced under both abnormal and normal conditions. A short table from a 1990 review by Milton Schlesinger makes the latter point (Table 5.1).
Table 5.1
Heat-shock protein inducers (after Schlesinger, J. Biol. Chem. 265: 12111, 1990)
Environmental stress conditions | Pathological stress conditions | Normal conditions |
|---|---|---|
Heat | Microbial infections | Cell cycle |
Heavy metals | Tissue trauma | Embryonic development |
Organics | Genetic lesions | Cell differentiation |
Oxidants | Hormonal stimulation | |
Microbial growth |
Those discoveries led John Ellis to reintroduce the concept of “molecular chaperones” in 1987 following a suggestion to that effect by Hugh Pelham in a 1986 paper. In that paper, Pelham summarized the possible role of these proteins in addition to a role in assembly. Specifically they (1) could assist in handling denatured or abnormal proteins’ (2) disrupt hydrophobic aggregates through their use of ATP, and (3) become upregulated in response to overloading of the protein degradation system.
5.1 Macromolecular Crowding Is Present in the Cell
The cellular environment is a crowded place; the fraction of the cellular volume occupied by dry matter is roughly 30 %, most of which is composed of proteins. That point is illustrated in Fig. 5.1 in which portions of typical eukaryotic and prokaryotic cells are shown using realistic representations of the resident macromolecule shapes and sizes. Crowding results in the steric inhibition of one macromolecule by another. One obvious consequence of the excluded volume is that it reduces diffusion rates. Beyond that, the effects of crowding are both less obvious and more interesting. Several studies have been carried out of how crowding affects protein folding. One conclusion is that crowding promotes the folding of globular proteins and enhances their refolding capabilities. It does so by reducing the conformation freedom of the unfolded chain far more than that of the compact native state, thus increasing the stability of the latter.
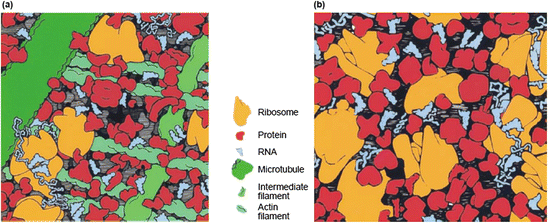
Fig. 5.1
Macromolecular crowding . Typical cytoplasm (a) in a eukaryotic cell and (b) in a bacterial (E. coli) cell. Squares depict the face of a cube of cytoplasm 100 nm in length on a side (from Ellis Curr. Opin. Struct. Biol. 11: 114 © 2001 Reprinted by permission from Elsevier)
The situation for intrinsically disordered protein s discussed in the last chapter is more complex and depends on the structural details and the ruggedness of the energy landscape . Significantly, it does not appear to impede the ability of these proteins to carry out their cellular tasks. Lastly, while crowding can enhance the speed at which a polypeptide chain can collapse, it can also increase the rate of aggregate formation especially of larger or more complex proteins that fold slowly, possess kinetic trap s, and/or do not rapidly bury hydrophobic patches in their interior.
A broad spectrum of tasks, all related to maintenance of protein health, that is, to proteostasis , is performed by molecular chaperones. As noted by Pelham and Ellis some time ago, molecular chaperones assist in the folding of nascent polypeptide chains in the crowded cellular environments. These chains are susceptible to forming inappropriate contacts with other proteins and with other domains of the same chain. Acting further downstream other molecular chaperones help to refold misfolded proteins, disaggregate proteins that have clumped together, and direct proteins that cannot be recovered to either the UPS or, if unavailable, to alternate disposal pathways. They also unfold proteins that have to cross intracellular membranes and refold them once they have transited through the membranes.
Not all proteins require the assistance of molecular chaperones. The fast folders, those proteins that fold rapidly through two-state kinetics in microseconds to milliseconds, do not, but others cannot fold without their help. Kinetic trap s are a key impediment to proper folding and for these proteins the assistance of chaperones is essential. As illustrated in Fig. 5.2 one way the chaperones promote folding is by destabilizing the kinetic trap s.
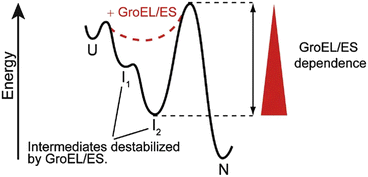
Fig. 5.2
Chaperone modification of the energy landscape for folding. GroEL/ES function destabilizes kinetic trap s, thereby enabling a protein to exit folding intermediates and reach its native state (from Kerner Cell 122: 209 © 2005 Reprinted by permission from Elsevier)
The ubiquitin-proteasome system and molecular chaperones occupy the upper layer of a multilayered defense system that protects against potentially toxic proteins. Molecular chaperones assist in folding of nascent polypeptide chains, the refolding of misfolded proteins, and the unclumping of aggregated ones. Any remaining malformed protein is directed either to the ubiquitin-proteosome system or undergo autophagic transport to lysosomes. Importantly, if these systems fail cells sequester the misfolded/aggregated/amyloidogenic proteins in inclusion bodies , the amyloidogenic deposits discussed in earlier chapter, and may even ship them out of the cell as will be discussed later in the text.
Inclusion bodies such as Lewy bodies , Pick bodies, and Bunina bodies contain ubiquitinated deposits of the misfolded proteins along with molecular chaperones and other components of the protein quality control system. In the remainder of this chapter and in the following one, the protein quality control system will be examined. One of the features that may characterize the proteins that aggregate and collect in IBs is that they are found at concentrations that are too high. Recall from the discussion of β2-microglobulin at the end of the last chapter that expression levels are important, and there is a need to maintain aggregation-prone proteins at low levels. The machinery responsible for the maintenance of proper concentration levels will be examined in the later, disease-specific chapters.
5.2 Ubiquitination Tags Proteins for Degradation by Proteosomes
Ubiquitin is a small protein, consisting of 76 amino acid residues with a mass of 8.5 kDa. It gets its name because of its ubiquitous presence in the cytosol and nucleus of bacteria, yeasts, higher plants, and animal cells. It was discovered in 1975 by Goldstein and coworkers and given the provisional name UBIP (ubiquitous immunopoietic polypeptide). The reversible attachment of this molecule to proteins has emerged as a major theme in cellular signaling since its role in degradation of short-lived species was first explored in the early 1980s by Ciechanover, Hershko, and Rose. The importance of this covalent modification was lent further support by the discoveries in just the last few years of the deubiquitinating enzymes.
Ubiquitin-dependent proteolysis is carried out in two stages. In the first step, the protein to be degraded is tagged by covalent attachment of a string of four or more ubiquitin molecules. In the second stage, the protein so-tagged is degraded within a large chambered multisubunit assemblage called the 26S proteasome. Three types of enzymes operating in a sequential fashion are responsible for attachment of the ubiquitin molecules to the target protein (Fig. 5.3). The first enzyme to act is an ubiquitin-activating, or E1 enzyme. It is followed by an ubiquitin-conjugating, or E2 enzyme, and finally by an ubiquitin-ligating, or E3 enzyme.
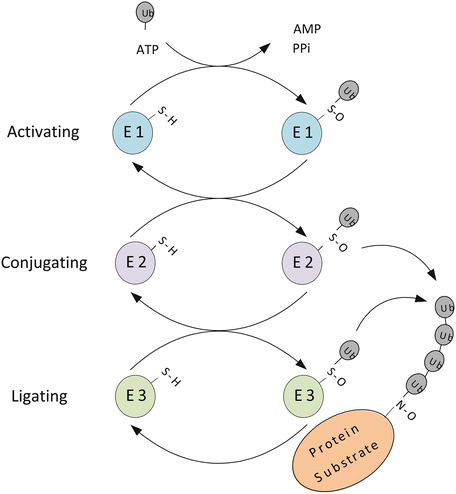
Fig. 5.3
Protein ubiquitination illustrating the three-stage process of ubiquitin attachment and chain growth. Ubiquitin possesses seven lysine residues, and can be conjugated on any of a number of these among which are K48 and K63. These two alternative sites for ubiquitination lead to different outcomes. In the case of K48 ubiquitination, the chain so produced leads to degradation of the tagged proteins by the 26S proteasome. In situations where K63 is the site for ubiquitination, the chain serves not as a tag for degradation but rather as a platform for assembly of complexes that activates downstream signaling
To begin the process the E1 catalyzes the attachment of an ubiquitin molecule to the sulfhydryl group on a catalytic cysteine residue within the E1. A high-energy thioester bond is formed between the sulfhydryl group and the carboxyl group of an ubiquitin C-terminus glycine residue. This is done in an ATP-dependent way as depicted in Fig. 5.3. Next, the E2 catalyzes the transfer of the ubiquitin protein from the E1 to an E2 catalytic cysteine residue to which it is again linked by means of a thioester bond. Lastly, the E3 catalyzes the transfer of the ubiquitin to an amino group of a side chain substrate lysine residue and to the growing multiubiquitin chain. Transfer to the substrate can occur either using the E3 as a transfer intermediate or directly from the E2. The ubiquitin-handling enzymes, the E1s, E2s, and E3s, form a hierarchy. A small number of E1s interact with a larger number of E2s and these interact with a yet larger number of E3s. The E3s confer substrate specificity to the degradation process acting either by themselves or together with the E2s.
5.3 Proteasomes Are Proteolytic Machines that Cleave the Tagged Proteins into Small Pieces
The 20S and 26S proteasomes are cylindrical-shaped proteolytic machines that degrade intracellular proteins. The 20S core unit (Fig. 5.4) is composed of four heptameric rings stacked one on top of the other with a total molecular mass of 750 kDa. The two inner rings consist of seven different β-subunits and the two outer rings contain seven different α-subunits. The β-subunits comprise the catalytic core of the proteasome while the α-subunits have a structural role and assist in controlling the entrance and exit of the substrate protein from the inner chamber. Three catalytic β-subunits positioned on the inner surface of each of the catalytic rings are responsible for cleaving peptide bonds. These units chop the client proteins into short peptide fragments ranging in size from 3 to 15 amino acid residues. These fragments are further degraded into amino acids by peptidases outside the proteasome.
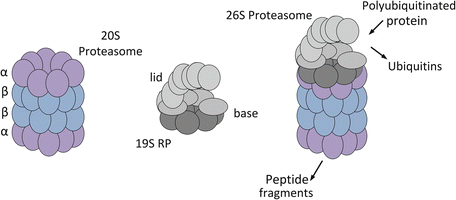
Fig. 5.4
Schematic diagrams of the 20S and 26S proteasomes based on electron microscopy and X-ray crystallography data. 20S proteasome: The outer α-subunits are shown in purple and the inner β subunits are depicted in blue. 19S regulatory particle (RP): The hexameric AAA ATPase ring that forms the base is shown in dark grey; the four Rnp subunits that belong to the base as well and the nine or so Rnps that comprise the lid are depicted in light grey. 26S proteasome: The structure shown is of a 20S proteasome capped by a single 19S RP
The 26S proteasome consists of a 20S catalytic core plus one or two 19S regulatory particles (RPs) that cap one or both ends of the core unit. The 19S RP contains two types of subunits—regulatory particles of AAA-ATPases (Rpts) and regulatory particles of non-ATPases (Rpns). The base subcomplex contains the six Rpts, which form a ring, plus four ancillary Rnps. The lid contains nine or more Rnps for a total of at least 19 subunits resulting in a 26S proteasome with a molecular mass of approximately 2 MDa. The proteolytic core chops substrate proteins in a nonspecific manner, but to accomplish this task the proteins must be inserted in an unfolded form through the narrow opening of the barrel-shaped 20S core. The lid of the 19S RP removes the ubiquitins from the protein. The base recognizes polyubiquitinated proteins, and prepares the protein for degradation by trapping and unfolding it. It then gates open the pore to allow entry of the unfolded protein into the 20S degradation chamber.
As just noted, in order for the 26S proteasome to degrade client proteins it must first unfold them and then feed the chain into the proteolytic chamber. In the 26S proteasome, the base of the 19S RP contains a hexameric AAA-ATPase, an unfoldase that handles the first task. The 20S proteasome, the peptidase, is then able to carry out the second task, degradation of the polypeptide chain. Shown in Fig. 5.5 is an example of the sequential action by ClpX(P), a heavily studied molecular machine that helps maintain protein quality control by degrading unwanted and misfolded proteins. In this example, the hexameric AAA-ATPase, ClpX, unfolds proteins, which are then fed into a double-ringed proteolytic chamber, ClpP. This molecular machine is present in bacteria and in mitochondria where proteins bearing a degradation tag, or degradon, are targeted for destruction. In these machines, like the proteasome, energy supplied by ATP hydrolysis is converted into mechanical work.
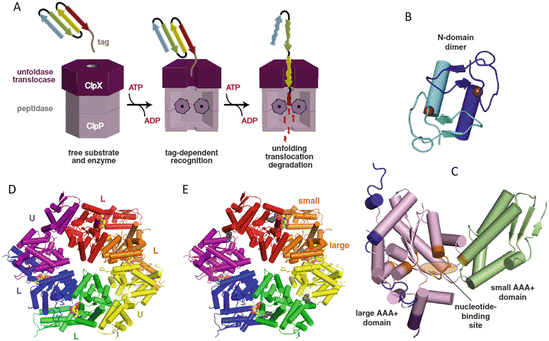
Fig. 5.5
The ClpXP molecular machine . (a) Schematic representation of the sequential unfolding and degradation operations. (b) Domain structure of ClpX N-domain dimer. (c) Domain structure of the AAA+ large and small domains within a subunit with L and U denoting the nucleotide loadable and unloadable subunits. (d) ClpX hexamer colored by subunit. (e) ClpX hexamer colored by rigid-body units (from Baker Biochim. Biophys. Acta 1823: 15 © 2012 Reprinted by permission from Elsevier)
5.4 Hsp60 Family Chaperonins Encapsulate Proteins in Folding Cages
Molecular chaperones are among the most highly expressed proteins in human cells. Recent analyses of proteomic data have revealed that some 10 % of the total protein mass is composed of molecular chaperones. These proteins can be grouped into a number of families each designated by their molecular weights—Hsp60, Hsp70, Hsp90, and Hsp100 plus sHSP, a family of small heat-shock proteins. Each family specializes in a particular set of proteostasis tasks. The chaperones form proteostasis networks in which a member of one family will hand off a problematic client protein to a member of a second family for further triage or to a protease for destruction if unrepairable.
Chaperonins directly participate in the folding process, supplying energy through ATP hydrolysis to the substrate protein. The additional energy supplied to the protein enables that protein to surmount energy barriers and escape from kinetic trap s. The client protein is able to fold far more rapidly into its native state since it can avoid getting stuck in nonoptimal and nonfunctional states for long periods of time. The subunits that form the folding cage repeatedly bind and release the substrate protein. They disrupt the misfolded structures that are kinetically trapped, and release the protein in a less folded configuration through the use of mechanical stretching forces. At the end of each round of binding and release, the protein is free to continue to fold and search for its low-energy native state.
There are two kinds of chaperonins. Group I chaperonins of which Hsp60 and GroEl are typical members are found in bacteria, mitochondria, and chloroplasts. These chaperonins require the presence of a co-chaperone, Hsp10 (GroES in bacteria), which functions as a lid over the Hsp60 (GroEL). Group I folding chambers are built from 16 identical subunits arranged in two rings, one stacked on top of the other, and are widely referred to as “Anfinsen cage s”. The operation of the GroEL/GRoEs folding chamber is shown in Fig. 5.6. Group II chaperonins, of which TRiC [t complex polypeptide 1 (TCP1) ring complex] is perhaps the best known member, are found in archaea and the eukaryotic cytosol. As was the case for Group I chaperonins, the Group II chaperonins are composed of two rings stacked on top of one another. However, in this class, the subunits possess an additional segment that serves as a lid and they do not require the assistance of a co-chaperone. Also, the type-II chaperonin rings are built from eight different subunits, each with its own unique role.
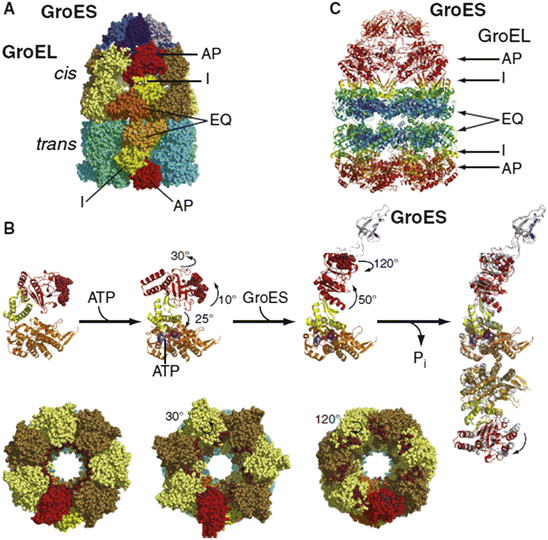
Fig. 5.6
Structure and changes in conformation that accompany ATP binding and hydrolysis. (a) Structure of GroEL/GroES complex. GroEL subunits are colored brown/yellow (cis-ring) and cyan/green-cyan (trans-ring); Equatorial (EQ), intermediate (I), and (AP) apical domains are colored orange, yellow, and red, respectively. GroES subunits are shaded in blue. (b) Conformational changes in a single GroEL subunit accompanying ATP binding and hydrolysis. (c) Debye-Waller factor (also called the thermal or b factor) for the GroEL/GroES complex colored blue-to-red for low-to-high. This factor provides a measure of the thermally driven conformational activity taking place within each structural element in the complex (from Mayer Mol. Cell 39: 321 © 2010 Reprinted by permission from Elsevier)
The encapsulation of a substrate protein within dedicated folding chambers serves several purposes. First, it sequesters the misfolded proteins from potential harmful interactions with other macromolecules in the crowded cellular environment. The folding chamber is, in fact, rather small, and this close confinement has a positive effect on the folding. As discussed in the preceding section on crowding, limiting the physical space that can be sampled by the polypeptide chain can by itself help collapse the protein and drive it towards the native state. Both of these effects are passive ones. In addition, the inner surface of the cage is lined with hydrophobic residues, and the subunits undergo large-scale movements driven by ATP hydrolysis. The chaperonin cage is much more than a passive chamber. Instead, it functions as a molecular machine that applies forces to the misfolded protein and does work on the proteins through repeated cycles of binding and release. Mechanistically, if these cycles are short, on the order of 1 s, the process can be regarded as occurring through iterative annealing. If, on the other hand, the cycles take longer, ~10–15 s, then the process may be thought of as one of forced unfolding.
5.5 Hsp70 Chaperones Maintain Cellular Proteostasis
Hsp70 family members assist in the folding of nascent proteins and proteins that have been exposed to cellular stresses resulting in their partial unfolding. They bind hydrophobic patches on newly synthesized chains, and bind and help transport proteins across organelle membranes and into the endoplasmic reticulum and mitochondria. They not only assist newly synthesized proteins in their folding to the native stage, but also prevent their aggregation and, if aggregation occurs, they help solubilize and refold the clumped proteins. For those proteins presenting difficulties, the Hsp70s will hand off proteins either to (1) Hps60s that form Anfinsen cage s to further refold kinetically trapped and difficult to fold proteins, or to (2) Hsp90s to maintain their stability and solubility and enable then to carry out their cellular functions. Alternatively, if the damage to the proteins is unrecoverable the Hsp70s will convey/send them to the UPS or via autophagy to lysosomes for degradation.
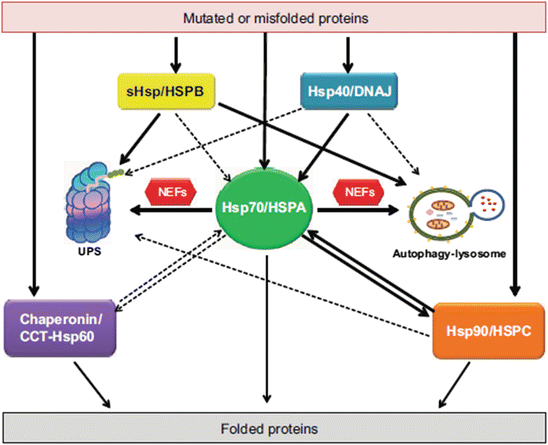
The aforementioned families of molecular chaperones operate as a highly integrated Hsp70/Hsp60/Hsp90 network. The Hsp70/HSPAs chaperones reside at the network center with Hsp60/chaperonins and Hsp90/HSPCs interconnected to them along with their co-chaperones and small heat-shock proteins (HSPBs). A schematic representation of this network is presented in Fig. 5.7. Several groups of co-chaperones assist in the above-mentioned activities. The Hsp70/Hsp90 organizing protein (HOP) facilitates the transfer from Hsp70s to Hsp90s. Chaperones belonging to the J-domain protein family such as Hsp40 and DnaJ (bacteria) provide substrate specificity to the Hsp70s; some J-proteins bind client proteins while others target the Hsp70s to the clients.
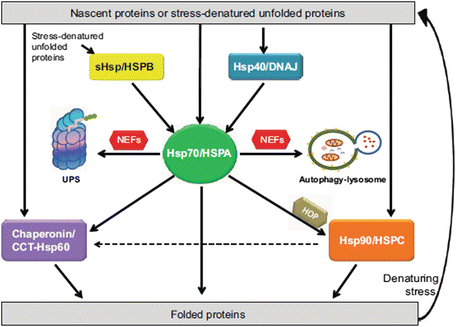
Fig. 5.7
The molecular chaperone network operating under acute stress conditions within the cell requiring their assistance in folding nascent proteins and in refolding stress-denatured unfolded proteins (from Kakkar Dis. Models Mech. 7: 421 © 2014 Reprinted by permission of the authors)
Hsp70 chaperones undergo multiple ADP/ATP cycles of binding and release. In this process, the J-domain proteins assist Hsp70 by working together with the substrate to lower the activation energy for hydrolysis. As depicted in Fig. 5.8, once bound, the substrate and co-chaperone stimulate hydrolysis of ATP leading to the closing of the substrate binding cavity about the substrate. In the next step, the ADP molecule is released to be rapidly replaced by another ATP molecule leading to opening of the substrate binding cavity, and the cycle is ready to be repeated. Another set of cofactors , the nucleotide exchange factors (NEFs), control the duration of the substrate binding by facilitating the exchange of ADP for ATP and triggering the release of the client protein.
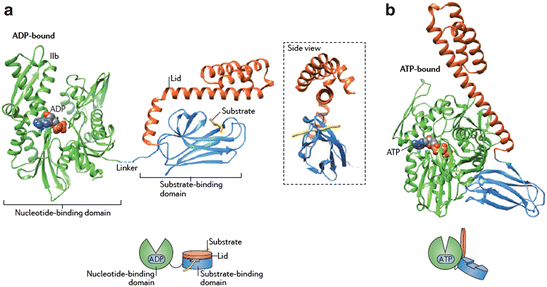
Fig. 5.8
Hsp70 domain structure and cycle of binding and release. (a) ADP-bound nucleotide-binding domain (NBD) is connected to the substrate-binding domain (SBD) plus lid by a flexible linker. (b) In the ATP-bound state the cleft in the NBD has narrowed and the lid and SBD undergo large conformational changes enabling release and exchange of portions of the substrate. Once hydrolysis occurs the lid closes back down onto the substrate and SBD (from Saibil Nat. Rev. Mol. Cell Biol. 14: 630 © 2013 Reprinted by permission from Macmillan Publishers Ltd)
5.6 Hsp90 Chaperones Stabilize Proteins
Each family of molecular chaperones has a distinct set of roles in preserving protein homeostasis. Members of the Hsp90 family play a key role in stabilizing proteins in nonnative intermediate states. To do so they form stable complexes with Hsp70, its co-chaperone Hsp40, and HOP. Once the Hsp90 binds and stabilizes its client, another cofactor , AHA1, triggers ATP hydrolysis resulting in client folding and release. Hsp90 chaperones play special roles in cellular signaling networks where they stabilize proteins that tend to be, at best, only weakly stable (or metastable) until they contact their signaling partners. Once these proteins establish contact they dissociate from the Hsp90s, and undergo stability-promoting conformational changes. The Hsp90 cycle of binding, stabilization, and activation is depicted in Fig. 5.9.
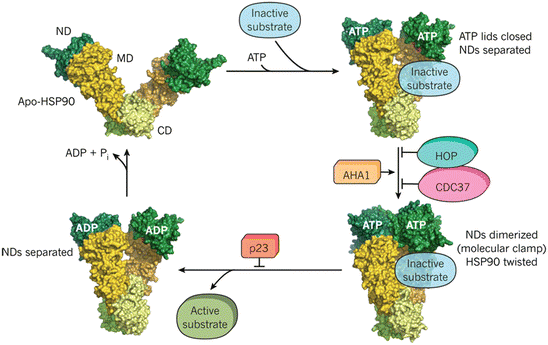
Fig. 5.9
The Hsp90 cycle. Upper-left panel: Hsp90 contains an N-terminal domain (ND), middle domain (MD), and C-terminal domain (CD), and functions as a dimer. Upper-right panel: ATP binding induces conformational changes and closure of the ATP lid. Lower-right panel: An ND dimer forms and the assemblage can function as a molecular clamp with its twisted subunits. Lower-left panel: ATP hydrolysis occurs resulting in activation and release of the substrate (from Hartl Nature 475: 324 © 2011 Reprinted by permission from Macmillan Publishers Ltd)
One of Hsp90s client proteins is the heat-shock stimulating factor Hsf1. This protein is the most conserved stress responder from yeasts to humans. It serves as a master transcription factor that binds one or multiple short sequences termed heat-shock elements (HSEs) located in promoters of a large number of target genes. Hsf1 not only regulates the transcription of heat-shock proteins but also regulates the transcription of proteins involved in related activities such as energy generation and protein trafficking. Under low-stress conditions Hsf1 is maintained in an inactive state through binding to Hsp90 and its cofactors . It detaches from Hsp90 in response to the buildup of ubiquitinated proteins indicative of protein folding-related stresses. Among the proteins upregulated by Hsf1 are Hsp70, Hsp40, the small heat-shock protein Hsp27, and Hsp90.
Interestingly and significantly, Hsp90 has many clients and it preserves not only partially-folded beneficial proteins but also misfolded, disease-causing ones. Among the Hsp90 clients are mutated proteins contributing to all of the hallmarks of cancer. In the case of neurodegeneration, something similar occurs with regard to preservation of aggregates of the primary contributors in each of the disease classes. This has given rise to the hope that by developing Hsp90 inhibitors, Hsf1 would be released, as it would in the case of cellular stresses, and upregulate transcription of the beneficial Hsp70s and Hsp40s.
5.7 Molecular Chaperones Handle Difficult-to-Treat Misfolded, Intrinsically Disordered, and Amyloidogenic Proteins
The handling of misfolded/intrinsically disordered, amyloidogenic proteins such as Aβ, α-synuclein, and Htt is especially challenging to the chaperone network. These proteins do not respond easily to refolding endeavors. Instead, they need to be sent either to the UPS or to the autophagic-lysosomal system for disposal. In these situations, the small heat-shock proteins send badly folded proteins directly to the degradation systems and not to the Hsp70s. This alternative routing by the HSPBs is depicted in Fig. 5.10. In addition, it is surmised that the Hsp40s/DNAJ co-chaperones may do the same. Proteostasis is maintained in these instances by rapidly disposing difficult-to-handle misfolders before they form toxic oligomer s and large potentially dangerous aggregates. A comparison of Fig. 5.10 to 5.7 shows that the situation is far more complex in the case of amyloidogenic and other difficult-to-handle proteins.