Rasmussen’s Encephalitis (Chronic Focal Encephalitis)
François Dubeau
Frédérick Andermann
Heinz Wiendl
Amit Bar-Or
Introduction
Chronic encephalitis is a relentlessly progressive disorder of childhood, associated with hemispheric atrophy, severe intractable focal epilepsy, intellectual decline, and hemiparesis. The etiology of this disorder remains unknown. Neuropathologic features described in the surgical specimens show characteristics of inflammatory changes including perivascular and leptomeningeal lymphocytic infiltration, microglial nodules, astrocytosis, neuronal degeneration, and spongy degeneration. There are variants of this syndrome with regard to age at onset, staging, localization, progression, and outcome. Treatment options are limited: Antiepileptic drugs (AEDs) usually show no significant benefit, and immunotherapy trials, undertaken mostly after the 1990 s, showed modest transient improvement in symptoms and disease progression in some patients. Only surgery, and specifically hemispherectomy, seems to produce persistent relief of seizures and functional improvement.
Historical Perspective
Dr. Theodore Rasmussen (Fig. 1) first described the disorder in 1958, and, together with Jerzy Olszewski and Donald Lloyd-Smith, published the clinical and histopathologic features of three patients with focal seizures due to chronic focal encephalitis.119 The original proband, F.S., was referred in 1945 to Dr. Wilder Penfield by Dr. Edgar Fincher, chief of neurosurgery at Emory University in Atlanta, GA, because of intractable right-sided focal motor seizures starting at 6 years of age.120 The child developed a right hemiparesis and underwent, between 1941 and 1956, three surgical interventions (two at the Montreal Neurological Hospital and Institute [MNHI]) at 7, 10, and 21 years in an attempt to control the evolution of the disease. In the first chapter of the monograph on chronic encephalitis published by Dr. Frederick Andermann in 1991, Dr. Rasmussen reported a letter of Dr. Fincher to Dr. Penfield (dated 1956) urging him to consider a more extensive cortical excision and concluded, “I note in your discussion that you list the cause as unknown, but if this youngster doesn’t have a chronic low-grade encephalitic process which has likely, by now, burned itself out, I will buy you a new hat.” The last intervention was a left hemispherectomy performed by Dr. Rasmussen, and histology showed sparse perivascular inflammation and glial nodules. W. G. remained seizure free until his last follow-up (Fig. 2B). He had a mild intellectual handicap and a fixed right hemiplegia. He developed hydrocephalus as a late complication of the surgical procedure and required a shunt. Dr. Penfield, who was consulted in this case, remained skeptical of the postulate that the syndrome was a primary inflammatory disorder, but raised most of the issues that continue to be debated: If it is an encephalitic process, would it not involve both hemispheres? Is the encephalitic process the result of recurrent seizures due to a small focal lesion in one hemisphere? Why it is that epileptic seizures are destructive in one case and not in another? Dr. Rasmussen himself recognized that Fincher’s 1941 diagnosis of chronic encephalitis in FS’s case was made 14 years before case two of the original 1958 report (Fig. 2).119 The story does not say, however, whether Dr. Penfield had to provide his colleague and friend Dr. Fincher with a new hat.5
This entity, later recognized as “Rasmussen’s encephalitis (RE),” became the subject of extensive discussion in the literature, initially debating the best timing for surgery and best surgical approaches, and, more recently, the etiology and pathogenesis of this unusual and enigmatic disease. A large number of publications can be found in the literature, and two international symposia were held in Montreal, in 1988 and in December 2002, and one in Vienna, in June 2004.21 The interest in this disease was initially driven by the severity and inescapability of its course, which rapidly led to its description as a prototype of “catastrophic childhood epilepsy.”
Physicians and scientists became interested by the unusual pathogenesis and evolution of the syndrome and are now trying to reconcile the apparent focal nature of the disease with the postulated viral and autoimmune etiologies that may or may not be mutually exclusive. This chapter updates a number of issues regarding RE, particularly the putative mechanisms of the disease, the variability of the clinical presentations, and the indications and rationale of new medical therapies, such as immunomodulation and receptor-directed pharmacotherapy.
Epidemiology
There are no data available regarding the incidence of RE in different populations. As the disorder has been increasingly recognized, reports and verbal communications from all parts of the world have emerged. This indicates that RE exists in every area, with the reports depending on the presence of pediatric neurologists and epileptologists. There are, however, no clusters of the disease in any particular region or population.
 FIGURE 1. Theodore B. Rasmussen, Director Emeritus of the Montreal Neurological Institute. |
Etiology and Pathogenesis of Rasmussen’s Encephalitis
The etiology and pathogenesis of RE remain unknown. Typical histologic features reported in surgical or autopsy specimens involve perivascular lymphocytic cuffing, microglial proliferation and nodule formation, neuronal loss, and gliosis in the affected
hemisphere (Fig. 3). The microglial nodules are associated with frequent nonspecific neuronophagia, and occur particularly near perivascular cuffs of lymphocytes and monocytes. There is limited evidence of spongiosis, which is not as widespread as in the true spongiform encephalopathies. Lesions tend to extend in a confluent rather than a multifocal manner. Finally, the main inflammatory changes are found in the cortex, and their intensity is inversely correlated with disease duration with slow progress toward a “burnt-out” stage.23,122 Three putative immune-mediated (inflammatory) mechanisms, which are not mutually exclusive, have been proposed to explain the initiation and unusual evolution of this rare clinical syndrome: (a) viral infections directly inducing central nervous system (CNS) injury, (b) a viral infection of the CNS that triggers a secondary autoimmune CNS process, and (c) a primary autoimmune CNS process. It remains possible that RE has a noninflammatory origin, and that the observed inflammatory responses merely represent a reaction to another unknown primary injury (Fig. 4).
hemisphere (Fig. 3). The microglial nodules are associated with frequent nonspecific neuronophagia, and occur particularly near perivascular cuffs of lymphocytes and monocytes. There is limited evidence of spongiosis, which is not as widespread as in the true spongiform encephalopathies. Lesions tend to extend in a confluent rather than a multifocal manner. Finally, the main inflammatory changes are found in the cortex, and their intensity is inversely correlated with disease duration with slow progress toward a “burnt-out” stage.23,122 Three putative immune-mediated (inflammatory) mechanisms, which are not mutually exclusive, have been proposed to explain the initiation and unusual evolution of this rare clinical syndrome: (a) viral infections directly inducing central nervous system (CNS) injury, (b) a viral infection of the CNS that triggers a secondary autoimmune CNS process, and (c) a primary autoimmune CNS process. It remains possible that RE has a noninflammatory origin, and that the observed inflammatory responses merely represent a reaction to another unknown primary injury (Fig. 4).
The observation of inflammatory responses found within the lesions of RE has led to multifaceted approaches to uncover possible infectious or immune-mediated (humoral or cellular) etiologies. In general, epidemiologic studies have not been able to identify clear genetic, geographic, seasonal, or clustering effect, and have failed to demonstrate any association between exposure to various factors, including viruses, and the subsequent development of RE. There appears to be no consistent increase in reports of pre-existent febrile convulsions, nor an association with an infectious illness preceding or associated with the development of RE. Serologic studies to detect antecedent viral infection have been contradictory or inconclusive.7,8,43,49,77,99,101,115,121,151,154 The search for a pathogenic virus has so far mostly focused on the herpes virus family, and direct brain tissue analysis has also yielded inconsistent results.7,8,77,99,116 Presently, the role of an infectious agent and the viral hypothesis in the causation of RE remains, at best, uncertain. It should be noted, however, that a few patients were reported to improve with antiviral therapy.35,37,96,100
Studies of both systemic62,87,93,118,127,132,133 and cerebrospinal fluid (CSF) compartment immune responses still fail to indicate clear evidence of either ongoing or deficient immune reactivity.60 A primary role for pathogenic antibodies in the etiology of RE was proposed after Rogers et al.123 described that rabbits immunized with fusion proteins containing a portion of the GluR3 (glutamate receptor 3 subunit) receptor developed intractable seizures. On histopathologic examination, the brains of these animals exhibited changes characteristic of RE with perivascular lymphocytic infiltrates and microglial nodules. The subsequent finding of autoantibodies to GluR3 in the sera of some affected patients with RE led to the GluR3 autoantibody hypothesis of RE and allowed new speculation into disease pathogenesis. GluR3 autoantibodies may cause damage to the brain, and eventually epilepsy, by excitotoxic mechanisms. In the animal model, GluR3 autoantibodies appear to activate the excitatory receptor that leads to massive influx of ions, neuronal cell death, local inflammation, and further disruption of the blood–brain barrier, allowing entry of additional immune mediators.90,145 Another proposed mechanism suggests that GluR3 autoantibodies can cause damage by activating complement cascades that lead to neuronal cell death and further inflammation.70,158 These hypotheses prompted a number of open-label therapeutic attempts in order to modulate the immune system of patients, especially by removing or blocking the circulating factors presumably responsible for the disease.3,6,70,109,123,145,158 Among cases with no detectable anti-GluR3 antibodies, several were also described to respond well to immunosuppressive treatments.5,66,155 Other reports in several patients showed no response to plasma exchange.3,82 More recent work, however, has shown that anti-GluR3 antibodies are not specific for RE but can be detected in other neurologic disorders and particularly in non-RE patients with severe epilepsy. Since the sensitivity of detection is low for the RE population and the presence of GluR3 antibodies does not distinguish RE from other forms of epilepsy, the anti-GluR3 antibody test is not useful for RE diagnosis.12,95,155,159 It remains unclear whether GluR3 or other autoantibodies in various forms of epilepsy are actually responsible for the onset of the seizure disorder, whether their presence contributes to ongoing pathophysiology of an established syndrome, or whether they merely result as an epiphenomenon of an underlying degenerative or inflammatory process.19,54 Passive transfer of the disease into naïve animals remains unsuccessful so far, and additional animal models of this illness are lacking to finally corroborate the potential pathogenic role of these antibodies.
Various other autoantibodies against neural molecules have been described in RE: Autoantibodies against munc-18,163 neuronal acetylcholine receptor α-7 subunit,156 and NMDAAR2 A to 2D—specifically GluR epsilon2136—have been reported in a number of patients. Again, however, these autoantibodies were not present in all the RE patients, and they could also be detected in neurologic diseases other than RE. This indicates that none of the described autoantibodies is specifically associated with RE, and that a variety of autoantibodies to neuronal and synaptic structures can be found that may contribute to the inflammatory process, or possibly represent an epiphenomenon of an activated immune system.
It is noteworthy that careful analysis of antibody gene rearrangement in lesions of chronic encephalitis patients demonstrated local clonal expansion of antibody producing cells12; the exact role of these humoral immune elements in the pathogenetic cascade of RE remains elusive. In the report by Takahashi et al.,136 anti-GluR epsilon2 antibodies were present only in patients with epilepsia partialis continua (EPC; 15 subjects, including ten with histologically proven or clinical RE, three with acute encephalitis/encephalopathy, and two with nonprogressive EPC), and were directed primarily against cytoplasmic epitopes, suggesting the involvement of T-cell–mediated autoimmunity.
It is noteworthy that careful analysis of antibody gene rearrangement in lesions of chronic encephalitis patients demonstrated local clonal expansion of antibody producing cells12; the exact role of these humoral immune elements in the pathogenetic cascade of RE remains elusive. In the report by Takahashi et al.,136 anti-GluR epsilon2 antibodies were present only in patients with epilepsia partialis continua (EPC; 15 subjects, including ten with histologically proven or clinical RE, three with acute encephalitis/encephalopathy, and two with nonprogressive EPC), and were directed primarily against cytoplasmic epitopes, suggesting the involvement of T-cell–mediated autoimmunity.
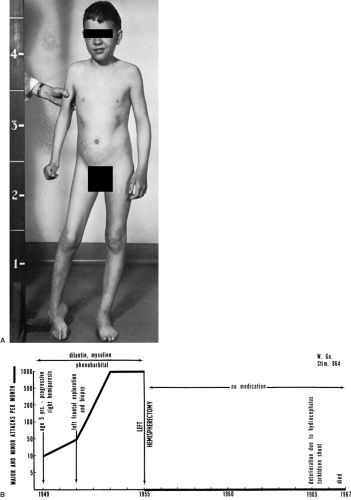 FIGURE 2. A: W. G., a boy with intractable seizures, right-sided hemiparesis, and hemiatrophy. This is the original patient described by Rasmussen. B: The course of W. G.’s illness. |
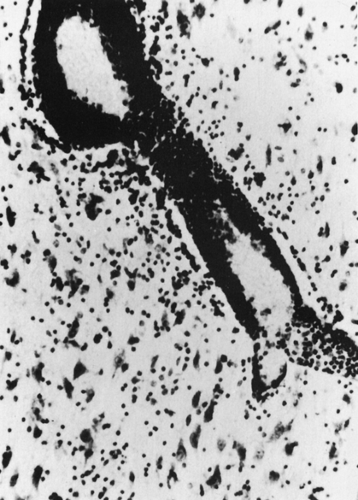 FIGURE 3. Pathologic findings in Rasmussen’s syndrome: Perivascular infiltrates, microglial nodules, and neuronophagia. |
Recent reports have indeed implicated a T-cell–mediated inflammatory response as another potential initiating or perpetuating mechanism in RE. Active inflammatory brain lesions contain large numbers of T lymphocytes,17 which appear to be recruited early within the lesions, implicating a T-cell–mediated immune response in the early evolution of the disease. Li et al.91 analyzed T-cell receptor expression in the lesions of patients with RE and found that the local immune response is characterized by restricted T-cell populations that have likely expanded from a small number of precursor T-cell clones, responding themselves to discrete antigenic epitopes. However, the nature of the antigens that trigger such a response is unknown. Recent work provides further credence to the hypothesis that a T-cell–mediated reaction, mainly consisting of cytotoxic CD8+ T-cell responses, may induce damage and apoptotic death of cortical neurons in RE.13,17,18 The demonstration of such cytotoxic T cells in close apposition to neurons suggests that RE might be a paradigm for a CD8-driven (auto) immune attack against neuronal structures. It is interesting to note that granzyme B, a toxic molecule secreted by CD8+ T cells upon interaction with a target, is capable of generating an antigenic epitope from the glutamate receptor.53 This observation might indicate a link between cellular and humoral immune components contributing to RE pathogenesis. In an attempt to integrate existing knowledge, several investigators13,17,19 have proposed a new scheme of pathogenesis. First, a focal CNS event initiates the process (e.g., infection, trauma, immune-mediated brain damage, even focal seizure activity) resulting in an immune reaction, involving antigen presentation in the CNS and entry of cytotoxic T lymphocytes into the CNS across the disrupted blood–brain barrier. Second, activated cytotoxic T lymphocytes attack CNS neurons while the inflammatory process, together with the release of cytokines, causes a spread of the inflammatory reaction and recruitment of more activated cytotoxic T lymphocytes. Third, the generation of potentially antigenic fragments, including GluR3, gives rise to autoantibodies, and may lead to an antibody-mediated “second wave of attack.” From an immunologic point of view, the typical adherence of the disease to one hemisphere still remains difficult to explain.
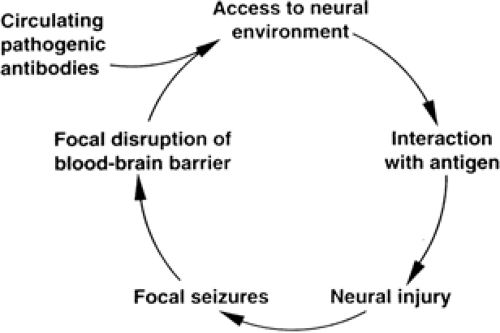 FIGURE 4. The “vicious cycle” hypothesis may explain Rasmussen’s encephalitis. Focal blood–brain barrier disruption facilitates entry of antibodies into the brain, with local neural injury and focal seizures. The focal seizures, in turn, cause focal transient blood–brain barrier disruption. |
Clinical Presentations
Typical Course of the Disease
In the early stages of the disease, the major issue is diagnosis. A combination of characteristic clinical, electrophysiologic, and imaging findings aids in the diagnosis. The 48 patients initially studied at the MNHI were collected over a period of 30 years and consisted mostly of cases referred from all over the world. Although now easier to recognize, this entity remains rare. During the last decade, an additional ten patients were studied in our institution, a small number compared to the 100 to 150 patients with intractable focal epilepsy due to other causes studied each year in our center. Typically, the disease starts in healthy children aged 1 to 13 years (mean age, 6.8 years) with 80% developing seizures before the age of 10 years.106 There is no difference in incidence between the sexes. In approximately half the patients, a history of infectious or inflammatory episode was described 6 months prior to the onset of seizures.
The first sign of the disease is the development of seizures. They are usually partial or secondarily generalized tonic–clonic seizures or status epilepticus (20% of the patients in the MNHI series presented with status epilepticus as the first manifestation). Early seizures could be polymorphic with variable semiology, but motor seizures are almost always reported. Other variable semiology of seizures with somatosensory, autonomic, visual, and limbic features has been described.58,106 The seizures rapidly become refractory, with little response to AEDs. EPC and other forms of focal motor seizures are particularly unresponsive to AEDs.39,63,114,142 We reviewed the AED therapy of 25 patients of the MNHI series and found no specific agent or combination therapy that appeared to be more effective or less toxic than other regimens.39 Our experience with newer AEDs in seven other patients with RE did not support improved effectiveness or tolerability for the new agents (personal data). The new antiepileptic agents levetira-cetam and topiramate may theoretically have a role in the treatment of RE, the first because of its efficacy in treating cortical myoclonus51,56 and the second because of its direct effect on glutamate receptors and release of N-methyl-D-glutamine.103 A variety of seizure types develop over time; the most common are focal motor and EPC (described in 56% of the patients in the MNHI series), with scalp electroencephalogram (EEG) patterns suggesting perirolandic onset. EPC is characterized by recurrent, asynchronous, and persistent (observed during wakefulness and sleep) myoclonias involving different muscle groups of the face, hand, or leg of one hemibody. Secondarily, generalized motor seizures are also described in many patients, but these appear to be easier to control with AEDs. Other less frequent types of motor seizures are a Jacksonian march (12%), posturing (25%), and versive movements of the head and eyes (13%), suggesting involvement of the primary, premotor, and supplementary motor areas. Drop attacks, however, are rare. Focal seizures with somatosensory (22% of patients), visual (16%), or auditory manifestations (2%) are also less frequent and appear later in the course of the disease, suggesting that the epileptogenic process has migrated from frontocentral and temporal regions to more posterior cortical areas, with a characteristic anteroposterior hemispheric march of the disease.106
Oguni et al.106 divided the progression of the disease into three stages: Stage 1, from the onset of the seizures and before the development of a fixed hemiparesis (3 months to 10 years, mean duration, 2.8 years); stage 2, from the development of a fixed hemiparesis (in 100% of the patients) to the completion of neurologic deterioration, including intellectual decline (in 85%), visual (49%) and sensory (29%) cortical deficits, and speech problems (dysarthria, 23%; dysphasia, 19%) dependent or independent of the burden of seizure activity (2 months to 10 years, mean duration, 3.7 years); and stage 3, stabilization of the condition in which further progression no longer occurs, and even the seizures may decrease in severity and frequency or, after some improvement, become again severe, continuous, and debilitating.
A more recent study by Bien et al.23 presented the clinical natural history of RE in parallel with the time course of brain destruction as measured by serial magnetic resonance imaging (MRI) in a series of 13 patients studied histologically. They separated the progression of the disease into prodromal, acute, and residual stages, comparable to the three stages of Oguni et al. Bien et al. distinguished two patterns of disease determined by the age at onset of RE: One with an earlier and more severe and rapidly progressive disorder starting during childhood (mean age at first seizure, 4.4 years; range, 1.6 to 6.4 years) and a second with a more protracted and milder course starting during adolescence or adult life (mean age at first seizure, 21.9 years; range, 6.4 to 40.9 years), the second pattern representing a variant of RE repeatedly described, particularly during the last decade.1,66,82,86,99
Clinical Variants of Rasmussen’s Syndrome
Rasmussen’s syndrome has been known for more than 50 years, and over 200 cases have now been reported.21 After the initial description, it became clear that the disease is clinically heterogeneous despite the pathologic hallmark of nonspecific chronic inflammation in the affected hemisphere. This heterogeneity may be explained by different etiologies (viral, viral- and non–viral-mediated autoimmune disease), by different reactions of the host’s immune system to exogenous or endogenous insults (age, genetic background, presence of another lesion, or “double pathology”), and by the modulating effect of a variety of antiviral, immunosuppressant, and immunomodulatory agents, or receptor-directed pharmacotherapy used in variable combinations and durations to treat these patients.
Atypical or unusual clinical features include early onset, usually before 2 years of age, with rapid propagation of the disease; bilateral cerebral involvement; relatively late onset during adolescence or adult life with slow progression; atypical anatomic location of the initial brain MRI findings; focal, protracted, or subcortical variants of RE; and double pathology. Also, two brothers developing bihemispheric disease early in
life and with a fatal outcome for one were reported by Silver et al.128 The brain biopsy of one of the brothers showed changes indistinguishable from the classical form of the disease.
life and with a fatal outcome for one were reported by Silver et al.128 The brain biopsy of one of the brothers showed changes indistinguishable from the classical form of the disease.
Bilateral Hemispheric Involvement
Usually the disease affects only one hemisphere, and most autopsy studies available confirmed unilateral cerebral involvement.122 Over time, however, there may be some contralateral ventricular enlargement and cortical atrophy attributed either to the effect of recurrent seizures and secondary epileptogenesis or to wallerian changes.85 Patients with definite bilateral inflammatory involvement are exceptional, and so far this has been described in no more than a dozen of them.31,37,67,99,128,135,141 Bilateral disease tends to occur in children with early onset (before 2 years). In these children, the disorder is usually fatal. Bilateral disease was also described in the late-onset adolescent or adult forms and is less severe in these patients. A small number had received high-dose steroids or an intrathecal antiviral agent, which suggested that early aggressive immunologic therapy may have predisposed to contralateral spread of the disease.37,135
Late-onset Adolescent and Adult Variants
A number of papers have reported the development of RE in adolescence or adult life as representing approximately 10% of the total number of patients with RE described in the literature over the last 40 years.1,23,32,50,59,66,71,82,84,86,89,99,104,131,146,147,157,164 In the MNHI series, 9 of 55 patients (16%) collected between 1945 and 2000 started to have seizures after the age of 12 years. The largest series, described by Hart et al.66 included 13 adults and adolescents collected from five centers. In comparison with the childhood form, late-onset RE has a more variable evolution,23,66 a generally more insidious onset of focal neurologic defects and cognitive impairment, and an increased incidence of occipital involvement (23% in the series of Hart et al. vs. 7% in children <12 years old in the MNHI series). Hemiparesis and hemispheric atrophy are often late and may not be as severe when compared with the more typical childhood form.23 Occasionally, however, the outcome in late-onset RE is similar to or worse than in children,59,99,104,131 but because of the generally more benign and protracted course, hemispherectomy seems less appropriate in this group of patients in whom neurologic deficits are usually less pronounced. Due to lack of brain plasticity in adults, the decision for hemispherectomy is even more complicated due to potential risk of new irreversible postoperative deficits.
Focal and Chronic Protracted Variants
There are rare reports of patients with RE whose seizures were relatively well controlled with AEDs or focal resections, and in whom the neurologic status stabilized spontaneously.66,71,88,164 Rasmussen had already suggested the existence of a “nonprogressive focal form of encephalitis.” With Aguilar, he reviewed 512 surgical specimens from 449 patients and found 32 cases with histologic evidence suggesting the presence of active encephalitis.1 Twelve patients demonstrated progressive neurologic deterioration compatible with RE, and 20 (4.4%) showed no or mild neurologic deterioration. In his review of patients who underwent temporal resections for intractable focal seizures, Laxer88 found five patients (3.8% of a series of 160 patients) with what he thought was a benign, focal, nonprogressive form of RE. These patients, children or adults, with no evidence of progression are indistinguishable clinically from those with refractory seizures due to other causes, including mesial temporal sclerosis.71,88
Delayed Seizure Onset Variant
Korn-Lubetski et al. recently reported on two children with the typical progressive clinical course and contralateral hemispheric atrophy characteristic for RE, but no clinical and EEG epileptic activity observed for several months.80 Both children had a brain biopsy (performed before seizure onset) confirming the clinical diagnosis. The focal seizures started in the first child 7 months after initial symptomatology and in the second after 6 months.
Basal Ganglia Involvement
Epilepsia partialis continua and other types of focal motor seizures are a common finding in patients with RE. Chorea, athetosis, and dystonia were infrequently described and may have been overlooked because of the preponderance of the epileptic disorder and of the hemiparesis. In 27 of the 48 patients of the MNHI series who had EPC, nine additionally had writhing or choreiform movements, and a diagnosis of Sydenham chorea was made in three of them early in the disease course.106 Matthews et al.97 described a 10-year-old girl with a 1-year history of progressive right-sided hemiparesis, EPC, and secondary generalized seizures. MRI showed diffuse cortical and subcortical changes, maximum in the perisylvian frontotemporoparietal area. At examination, she had choreic movements of the right arm and hand in addition to EPC. Tien et al.140 were the first to describe in an 8.5-year-old girl with intractable focal motor seizures and atrophy of the caudate and putamen, with abnormal high signals and severe left hemispheric atrophy. They interpreted these findings as the result of gliosis and chronic brain damage. Topçu et al.142 described a patient who developed hemidystonia as a result of involvement of the contralateral basal ganglia. The movement disorder appeared 3 years after onset of seizures. A rather typical subsequent evolution suggested RE. The movement disorder started during intravenous immunoglobulin (IVIg) and interferon therapy, and did not respond to anticholinergic drugs nor to a frontal resection. Ben-Zeev et al.,15 Koehn and Zupanc,79 Frucht,50 and, finally, Lascelles et al.87 each reported a case of RE whose clinical presentation was dominated by a hemidys-kinesia, with EPC in three of those patients, and progressive hemiparesis. Two cases showed selective frontal cortical and caudate atrophy on MRI, one developed progressive left basal ganglia atrophy and later focal frontotemporoparietal atrophy, and one had only pronounced right caudate, globus pallidus, and putamen atrophy. In the case of Frucht, IVIg dramatically improved both the hyperkinetic movements and the EPC, but the effect was transient, suggesting a common neuroanatomic mechanism or humoral autoimmune process. In a series of 21 patients with RE, Bhatjiwale et al.16 looked specifically at the involvement of the basal ganglia. Fifteen (71%) showed mild to severe basal ganglia involvement on imaging in three different patterns: Predominantly cortical in six cases, predominantly basal ganglia in six, and with both cortical and basal ganglia involvement in six. In five cases, the changes found in the basal ganglia were static, whereas in the others there was steady progression. The caudate nucleus was generally more prominently involved, usually in association with frontal atrophy. Five cases also showed putaminal involvement, always with temporoinsular atrophy. Interestingly, two of the six patients with prominent basal ganglia involvement had dystonia as a presenting feature. The authors postulated that the disease may proceed from different foci, including cases where RE seems to start in deep gray matter. The Italian Study Group recently described similar findings on Rasmussen’s encephalitis,30 which found basal ganglia atrophy in 9 of 13 patients studied. They suggested that atrophy of the basal ganglia represents secondary changes due to disconnection from the affected overlying frontal and insular cortex.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


