Chapter 8 REM Sleep*
Abstract
Rapid eye movement, or REM, sleep was discovered by Aserinsky and Kleitman in 1953.1 In a beautifully written paper that has stood the test of time, they reported that REM sleep was characterized by the periodic recurrence of rapid eye movements, linked to a dramatic reduction in amplitude from the higher-voltage activity of the prior non-REM sleep period, as seen on the electroencephalogram (EEG). They found that the EEG of subjects in REM sleep closely resembled the EEG of alert–waking subjects, and they reported that subjects awakened from REM sleep reported vivid dreams. Dement identified a similar state of low-voltage EEG with eye movements in cats.2 Jouvet then repeated this observation, finding in addition a loss of muscle tone (i.e., atonia) in REM sleep and using the term paradoxical sleep to refer to this state. The paradox was that the EEG resembled that of waking, but behaviorally the animal remained asleep and unresponsive.3–5 Subsequent authors have described this state as activated sleep, or dream sleep. More recent work in humans has shown that some mental activity can be present in non-REM sleep but has supported the original finding that linked our most vivid dreams to the REM sleep state. However, we cannot assume that any person or animal who has REM sleep also dreams. Lesions of parietal cortex and certain other regions prevent dreaming in humans, even in individuals continuing to show normal REM sleep as judged by cortical EEG, suppression of muscle tone, and rapid eye movements.6 Children younger than 6 years, who have larger amounts of REM sleep than adults, do not typically report dream mentation, perhaps because these cortical regions have not yet developed.7 The physiologic signs of REM sleep in both the platypus, the animal showing the most REM sleep,8 and a related monotreme, the short-nosed echidna,9 are largely restricted to the brainstem, in contrast to their propagation to the forebrain in adult placental and marsupial mammals. These findings make it questionable whether all or any nonhuman mammals that have REM sleep, all of which have cortical regions whose structure differs from that of adult humans, have dream mentation.
Characteristics of REM Sleep
The principal electrical signs of REM sleep include a reduction in EEG amplitude, particularly in the power of its lower-frequency components (Fig. 8-1). REM sleep is also characterized by a suppression of muscle tone (atonia), visible in the electromyogram (EMG). Erections tend to occur in males.10 Thermoregulation (e.g., sweating and shivering) largely ceases in most animals, and body temperatures drift toward environmental temperatures, as in reptiles.11 Pupils constrict, reflecting a parasympathetic dominance in the control of the iris.12 These changes that are present throughout the REM sleep period have been termed its tonic features.
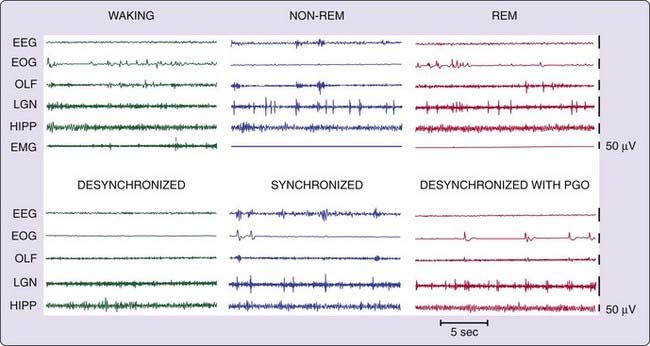
Also visible are electrical potentials that can be most easily recorded in the lateral geniculate nucleus of the cat.13 These potentials originate in the pons, appear after a few milliseconds in the lateral geniculate nucleus, and can be observed with further delay in the occipital cortex, leading to the name ponto-geniculo-occipital (PGO) spikes. They occur as large-amplitude, isolated potentials 30 or more seconds before the onset of REM sleep as defined by EEG and EMG criteria. After REM sleep begins, they arrive in bursts of three to ten waves, usually correlated with rapid eye movements. PGO-linked potentials can also be recorded in the motor nuclei of the extraocular muscles, where they trigger the rapid eye movements of REM sleep. They are also present in thalamic nuclei other than the geniculate and in neocortical regions other than the occipital cortex.
In humans, rapid eye movements are loosely correlated with contractions of the middle ear muscles of the sort that accompany speech generation and that are part of the protective response to loud noise.14 Other muscles also contract during periods of rapid eye movement, briefly breaking through the muscle atonia of REM sleep. There are periods of marked irregularity in respiratory and heart rates during REM sleep, in contrast to non-REM sleep, during which respiration and heart rate are highly regular. No single pacemaker for all of this irregular activity has been identified. Rather, the signals producing twitches of the peripheral or middle ear muscles may lead or follow PGO spikes and rapid eye movements. Bursts of brainstem neuronal activity may likewise lead or follow the activity of any particular recorded muscle.15–17 These changes that occur episodically in REM sleep have been called its phasic features.
As we will see later, certain manipulations of the brainstem can eliminate only the phasic events of REM sleep, whereas others can cause the phasic events to occur in waking; yet other manipulations can affect tonic components. These tonic and phasic features are also expressed to varying extents in different species, and not all of these features are present in all species that have been judged to have REM sleep.18
REM Generation Mechanisms
Technical Considerations
Inactivation of Neurons by Lesions, Inhibition, Antisense Administration, or Genetic Manipulation
It will soon be possible to acquire mutant mice in which any one, or several, of more than 20,000 genes are inactivated, to the extent that such deletions are not lethal. Investigation of two mutants19,20 led to major insights into the etiology of human narcolepsy.21–23 Techniques for the postnatal inactivation of genes permit investigation of gene deletions without the developmental effect of these deletions. They can also be used for investigation of the effects of gene inactivation in particular brain regions. A similar inactivation can be achieved by localized microinjections of antisense. Many if not most such mutants can be expected to have some sleep phenotype, such as increases or decreases in total sleep or REM sleep time, altered sleep rebound, altered responses of sleep to environmental variables, and altered changes in sleep with development and aging. The same interpretive considerations long appreciated in lesion studies apply to the interpretation of manipulations that inactivate genes or prevent gene expression, with the additional possibility of direct effects of genetic manipulation on tissues outside the brain.
Activation of Neurons by Electrical or Chemical Stimulation, Gene Activation, or Ion Channel Manipulation
Sites identified by lesion or anatomic studies can be stimulated to identify their roles in sleep control. Older studies used electrical stimulation and were successful in identifying the medial medulla as a region mediating the suppression of muscle tone,24 and the basal forebrain as a site capable of triggering sleep.25 Electrical stimulation is clearly an aphysiologic technique, involving the forced depolarization of neuronal membranes by ion flow at a frequency set by the stimulation device, rather than by the patterned afferent impulses that normally control neuronal discharge. For this reason, it has been supplanted for many purposes by administration of neurotransmitter agonists, either by direct microinjection or by diffusion from a microdialysis membrane that is placed in the target area and perfused with high concentrations of agonists.
Because it is far easier to inject a substance than to collect and quantify physiologically released ligands, there have been many studies implying that various substances are critical for REM sleep control based solely on microinjection. These results must be interpreted with caution. For example, hypocretin is known to depolarize virtually all neuronal types. It should therefore not be surprising to find that hypocretin microinjection into arousal systems such as the locus coeruleus produces arousal,26 that microinjection of hypocretin into sites known to control feeding increases food intake,27 that injection into regions known to contain cells that are waking active increase waking,28 that injection into regions known to contain cells selectively active in REM sleep will increase the occurrence of this state,29,30 that injection into regions known to facilitate muscle tone will increase tone, that identical injections into regions known to suppress tone will decrease tone,31 and that intracerebroventricular injection of hypocretin can increase water intake32 and can activate other periventricular systems.29 Such types of findings do not by themselves demonstrate a role for hypocretin (or any other neurotransmitter) in the observed behavior. It is necessary to obtain data on the effects of inactivation of, for example, hypocretin or hypocretin receptors, and to record evidence that indicates activity of hypocretin neurons at the appropriate times before seriously entertaining such conclusions.
Genetic manipulations enable activation of neurons or nonneuronal cells of a particular type. A recent example of a genetic approach is the insertion of a light-sensitive ion channel into hypocretin cells using a lentivirus. Fiberoptic delivery of light could then be used to activate just these cells and determine the effect on sleep–waking transitions.33
Observation of Neuronal Activity
Recording the activity of single neurons in vivo can provide a powerful insight into the precise time course of neuronal discharge. Unit activity can be combined with other techniques to make it even more useful. For example, electrical stimulation of potential target areas can be used to antidromically identify the axonal projections of the recorded cell. Intracellular or juxtacellular34 labeling of neurons with dyes, with subsequent immunolabeling of their transmitter, can be used to determine the neurotransmitter phenotype of the recorded cell. Combined dialysis and unit recording or iontophoresis of neurotransmitter from multiple-barrel recording and stimulating micropipettes can be used to determine the transmitter response of the recorded cell, although it cannot be easily determined whether the effects seen are the direct result of responses in the recorded cell or are mediated by adjacent cells projecting to the recorded cell. Such distinctions can be made in in vitro studies of slices of brain tissue by blocking synaptic transmission or by physically dissociating studied cells, but in this case their role in sleep may not be easily determined.
Despite this mismatch of time courses, the tonic latency, a measure of how long before REM sleep–onset activity in a recorded cell changes, has been computed in some studies. Neurons purported to show a “significant” change in activity many seconds or even minutes prior to REM sleep onset have been reported. However, such a measure is of little usefulness, because at the neuronal level, the activity of key cell groups can best be seen as curvilinear over the sleep cycle, rather than changing abruptly in the way that activity follows discrete sensory stimulation. A major determinant of the tonic latency, computed as defined here, is the level of noise, or variability in the cell’s discharge, which affects the difficulty of detecting a significant underlying change in rate in a cell population. It is therefore not surprising that cell groups designated as executive neurons for REM sleep control on the basis of their tonic latencies were later found to have no essential role in the generation of REM sleep.35–37 The more appropriate comparison of the unit activity cycle to state control is to compare two different cell types to see what the phase relationship of the peaks or troughs of their activity is, under similar conditions. This kind of study is difficult, involving the simultaneous long-term recording of multiple cells, and it is rarely performed. Even in this case, a phase lead does not by itself prove that the “lead” neuron is driving activity seen in the “following” neuron, but it does indicate that the reverse is not the case. However, awakening is a process that can be studied in this way, because it can be elicited by stimuli, and it appears to be preceded by abrupt changes in the activity of many neuronal groups.38 A major advantage of unit recording approaches in the intact animal to understand sleep and other behavioral processes is their high level of temporal resolution.
Observation of the normal pattern of neurotransmitter release and neuronal activity can help determine the neurochemical correlates of sleep states. The natural release of neurotransmitters can be most easily determined by placing a tubular dialysis membrane 1 to 5 mm in length in the area of interest and circulating artificial cerebrospinal fluid through it. Neurotransmitters released outside the membrane will diffuse through the membrane and can be collected. Each sample is collected at intervals, typically ranging from 2 to 10 minutes. The collected dialysates can be analyzed by chromatography, radioimmunoassay, mass spectroscopy, or other means. The temporal resolution of this technique is typically on the order of a few minutes for each sample.39–41
Unit recording and dialysis approaches require a sharp research focus on a particular neurotransmitter or neuronal group. In contrast, histologic approaches can be used to measure the activity of the entire brain at cellular levels of resolution. The most popular such approach in animal studies labels the activation of immediate early genes. These genes are expressed when a neuron is highly active, and their expression is an early step in the activation of other downstream genes mobilizing the response of the cell to activation. The likelihood of such expression may vary between neuronal types and may not be detectable below some activity-dependent threshold. Activation of these genes can be detected by immunohistochemistry, most commonly by staining for the production of the Fos protein or the mRNA used to synthesize this protein.42 Neurons can be double-labeled to determine the transmitter they express, allowing investigators to determine, for example, whether histaminergic neurons in the posterior hypothalamus were activated in a particular sleep or waking state. Metabolic labels such as 2-deoxyglucose can also provide an indication of which neurons are active.42,43 Similar techniques using radioactive ligands in positron emission tomography (PET) studies can be used in living humans or animals. In vivo measurements of blood flow can be made throughout the brain with functional magnetic resonance imaging (fMRI). All of these techniques have in common an ability to make anatomically driven discoveries of brain region involvement in particular states, independent of specific hypotheses, thus leading to major advances in understanding. However, another common feature of these types of recording techniques is their very poor temporal and spatial resolutions compared with neuronal recording approaches. Fos activation can take 20 minutes or more. PET takes a similar amount of time, and fMRI can be used to observe events lasting on the order of 1 to 15 seconds. It cannot be known whther areas active during a particular state caused the state or were activated because of the state.
Transection Studies
The most radical types of lesion studies are those that slice through the brainstem, severing the connections between regions rostral and caudal to the cut. Sherrington43a discovered that animals, whose forebrain was removed after transecting the neuraxis in the coronal plane at the rostral border of the superior colliculus, showed tonic excitation of the “antigravity muscles” or extensors (Fig. 8-2, level A). This decerebrate rigidly was visible as soon as anesthesia was discontinued. Bard and Macht reported in 1958 that animals with decerebrate rigidity would show periodic limb relaxation.44 We now know that Bard was observing the periodic muscle atonia of REM sleep.
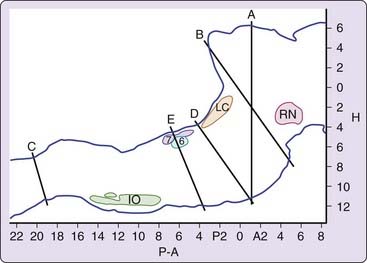
After the discovery of REM sleep in the cat,2 Jouvet found that this state of EEG desynchrony was normally accompanied by muscle atonia.4 Jouvet then examined the decerebrate cat preparation used by Sherrington and Bard, now adding measures of muscle tone, eye movement and EEG. One might have expected that REM sleep originated in the forebrain, but Jouvet found something quite different. When he recorded in the forebrain after separating the forebrain from the brainstem at the midbrain level (see Fig. 8-2, levels A or B), he found no clear evidence of REM sleep. In the first few days after transection, the EEG in the forebrain always showed high voltage, but when low-voltage activity appeared, the PGO spikes that help identify REM sleep in the intact animal were absent from the thalamic structures, particularly the lateral geniculate where they can be most easily recorded. Thus it appeared that the isolated forebrain had slow-wave sleep states and possibly waking, but no clear evidence of REM sleep.
An interesting feature of REM sleep in the decerebrate animal is that its frequency and duration varied with the temperature of the animal. In the decerebrate animal, the forebrain thermoregulatory mechanisms are disconnected from their brainstem effectors. Shivering and panting do not occur at the relatively small temperature shifts that trigger them in the intact animal. For this reason, if the body temperature is not maintained by external heating or cooling, it will tend to drift toward room temperature. Arnulf and colleagues45 found that if body temperature was maintained at a normal level, little or no REM sleep appeared. But if temperature was allowed to fall, REM sleep amounts increased to levels well above those seen in the intact animal. This suggests that REM-sleep facilitatory mechanisms are, on balance, less impaired by reduced temperature than are REM-sleep inhibitory mechanisms. Another way of looking at this phenomenon is that brainstem mechanisms are set to respond to low temperatures by triggering REM sleep, perhaps to stimulate the brainstem, and that high brainstem temperatures inhibit REM sleep. It is unclear whether this mechanism is operative in the intact animal where temperature shifts are within a much narrower range.
A further localization of the REM sleep control mechanisms can be achieved by examining the sleep of humans or animals in which the brainstem-to-spinal cord connection has been severed (see Fig. 8-2, level C). In this case, normal REM sleep in all its manifestations, except for spinally mediated atonia is present.46 Thus we can conclude that the region between the caudal medulla and the rostral midbrain is sufficient to generate REM sleep.
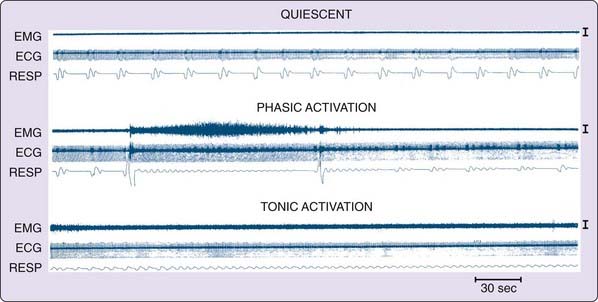
This approach can be continued by separating the caudal pons from the medulla (see Fig. 8-2, level D or E). In such animals, no atonia is present in musculature controlled by the spinal cord, even though electrical or chemical stimulation of the medial medulla in the decerebrate animal suppresses muscle tone.47 Furthermore, neuronal activity in the medulla does not resemble that seen across the REM–non-REM sleep cycle, with neuronal discharge very regular for periods of many hours, in contrast to the periodic rate modulation that is linked to the phasic events of REM sleep in the intact animal (Fig. 8-3).48 This demonstrates that the medulla and spinal cord together, although they may contain circuitry whose activation can suppress muscle tone, are not sufficient to generate this aspect of REM sleep when disconnected from more rostral brainstem structures.
In contrast, the regions rostral to this cut show aspects of REM sleep (Fig. 8-4, and see Fig. 8-1, bottom).49 In these regions, we can see the progression from isolated to grouped PGO spikes and the accompanying reduction in PGO spike amplitude that occurs in the pre-REM sleep period and the REM sleep periods in the intact animal. We also see increased forebrain unit activity, with unit spike bursts in conjunction with PGO spikes, just as in REM sleep.48,50
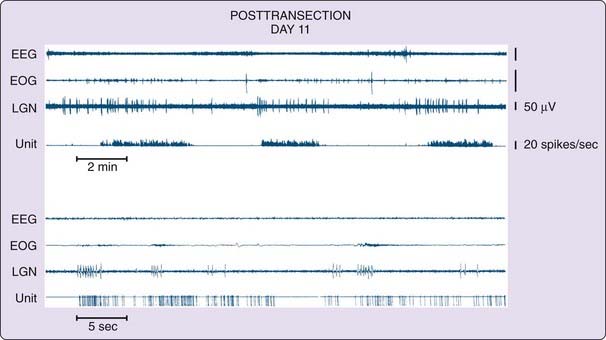
To summarize, this work shows that when pontine regions are connected to the medulla, atonia, rapid eye movements and the associated unit activity of REM sleep occur, whereas the medulla and spinal cord together, disconnected from the pons, are not sufficient to generate these local aspects of REM sleep. When the pons is connected to the forebrain, forebrain aspects of REM sleep are seen, but the forebrain without attached pons does not generate these aspects of REM sleep. Further confirmation of the importance of the pons and caudal midbrain comes from the studies of Matsuzaki,51 who found that when two cuts were placed, one at the junction of the midbrain and pons and the other at the junction of the pons and medulla, periods of PGO spikes could be seen in the isolated pons, but no signs of REM sleep in structures rostral or caudal to the pontine island.
However, it is also clear that the pons alone does not generate all the phenomena of REM sleep. Atonia requires the activation of motor inhibitory systems in the medulla.52 In the intact animal, forebrain mechanisms interact with pontine mechanisms to regulate the amplitude and periodicity of PGO spikes,53 which in turn are linked to the twitches and rapid eye movements of REM sleep. We know from cases of human REM sleep behavior disorder that the motor activity expressed in dreams is tightly linked to the imagery of the dream.54 Extrapolating to dream imagery in normal humans, one can hypothesize that because the structure of REM sleep results from an interaction of forebrain and brainstem mechanisms, the dream itself is not just passively driven from the brainstem but rather represents the result of a dynamic interaction between forebrain and brainstem structures.
Localized Lesion Studies
The transection studies point to a relatively small portion of the brainstem—the pons and caudal midbrain—as being critical for REM sleep generation. Further specification of the core regions can be achieved by destroying portions of the pons in an otherwise intact animal and seeing which areas are necessary and which are unnecessary for REM sleep generation. An early systematic study by Carli and Zanchetti in the cat55 and other subsequent studies emphasized that lesions of locus coeruleus56 and the dorsal raphe57 nuclei, or of simultaneous lesions of locus coeruleus, forebrain cholinergic neurons, and histamine neurons,58 did not block REM sleep. Carli and Zanchetti concluded that lesions that destroyed the region ventral to the locus coeruleus, called the nucleus reticularis pontis oralis or the subcoeruleus region, produced a massive decrease in the amount of REM sleep. In their studies, Carli and Zanchetti used the electrolytic lesion technique, in which a current is passed, depositing metal that kills cells and axons of passage. As cytotoxic techniques that allowed poisoning of cell bodies without damage to axons of passage came into use, these initial conclusions were confirmed and refined. It was shown that neurons in medial pontine regions including the giant cell region were not important in REM sleep control,52,59,60 as near-total destruction of these cells was followed by normal amounts of REM sleep as soon as anesthesia dissipated.36,61 However, lesions of the subcoeruleus and adjacent regions with cytotoxins did cause a prolonged reduction in the amount of REM sleep. According to one study, the extent of this loss was proportional to the percentage of cholinergic cells lost in subcoeruleus and adjacent regions of the brainstem of the cat.62 In rats, lesion or inactivation of the same region below the locus coeruleus (called the sublaterodorsal nucleus in the terminology of Swanson63) has been found to reduce REM sleep.64
Although large lesions may eliminate all aspects of REM sleep, small bilaterally symmetrical lesions in the pons can eliminate specific aspects of REM sleep. Lesions of lateral pontine structures allow muscle atonia during REM sleep. However, PGO spikes and the associated rapid eye movements are absent when lesions include the region surrounding the superior cerebellar peduncle of the cat (Fig. 8-5, top).65 This points to the role of this lateral region in the generation of PGO waves and some of the associated phasic activity of REM sleep.
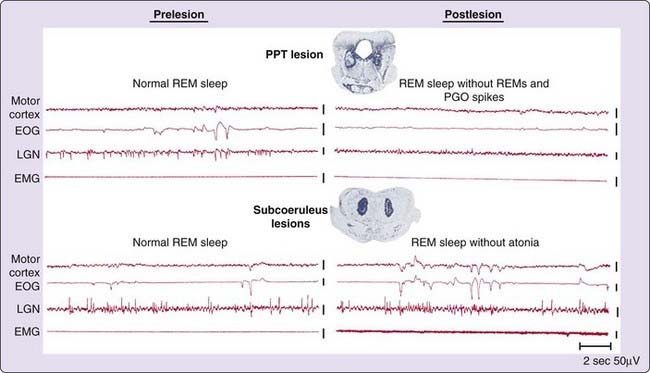
Small lesions confined to portions of the subcoeruleus regions identified as critical for REM sleep by Carli and Zanchetti, or to the medial medulla52 result in a very unusual syndrome. After non-REM sleep, these animals enter REM sleep, as indicated by lack of responsiveness to the environment, PGO spikes, EEG desynchrony, and pupil constriction. However, they lack the muscle atonia that normally characterizes this state (see Fig. 8-5, bottom).5,66 During REM sleep without atonia, these animals appear to act out dreams, attacking objects that are not visible, exhibiting unusual affective behaviors and ataxic locomotion. When they are awakened, normal behavior resumes. More recent studies have demonstrated that lesions of a system extending from the ventral midbrain to the medial medulla can cause REM sleep without atonia and that activation of this system can suppress muscle tone.52,67–69
This subcoeruleus region is under the control of midbrain regions. A midbrain region located just beneath and lateral to the periaqueductal gray (and called the dorsocaudal central tegmental field in the cat), appears to inhibit REM sleep by inhibiting the critical REM-on subcoeruleus neurons. Muscimol, a gamma-aminobutyric acid A (GABAA) receptor agonist, injected into this midbrain region, silences these cells and increases REM sleep, presumably by blocking the inhibition.70 The same phenomena have been observed when muscimol is injected into the corresponding region of the guinea pig71 and the rat (in the rat, this midbrain region has been called the deep mesencephalic nucleus).72 The midbrain region of the deep mesencephalic nucleus is the heart of the classic reticular activating system, shown to induce waking when electrically stimulated,73 and coma when lesioned.74 Both of these manipulations affect not only intrinsic neurons but also axons of passage.
Increasing the levels of GABA in the subcoeruleus region (also called the pontine oralis nucleus in the rat and cat) produces an increase in waking, rather than the increase in REM sleep seen with GABA injection into the midbrain regions indicated previously.75,76 This is another reminder that, despite the sleep-inducing effect of systemic administration of hypnotic medications, local manipulation shows that the effect of GABA on sleep and waking states varies across brain regions. Blocking GABA in the subcoeruleus has been reported to increase REM sleep in the cat.77
Stimulation Studies
The first study showing that stimulation could elicit REM sleep was carried our by George and colleagues.78 They found that application of the acetylcholine agonist carbachol to specific regions of the pons ventral to the locus coeruleus could elicit REM sleep in the cat. An impressive proof that a unique REM sleep–generation mechanism was being activated was the long duration of the elicited REM sleep periods. Microinjection of acetylcholine into this region in the decerebrate cat produces an immediate suppression of decerebrate rigidity. Later studies showed that, depending on the exact site, either REM sleep or just atonia in a waking state could be triggered by such stimulation.79–81 When stimulation was applied to the lateral regions whose lesion blocked PGO waves, continuous PGO spikes were generated even though the animal was not always behaviorally asleep.
Increased REM sleep has been reported in the rat after microinjection of cholinergic agonists into the subcoeruleus region,82–84 although this effect is certainly not as robust as it is in the cat.85
The first study demonstrating a role for glutamate in the control of REM sleep was done in the cat. We found that a profound suppression of muscle tone could be elicited by the injection of glutamate into the subcoeruleus region or into the ventral medullary region.47,86,87 Further work has demonstrated that the pontine cells in this inhibitory region receiving this cholinergic input use glutamate as their transmitter and project directly to glutamate-responsive regions of the medial medulla.86,88–90
Work in the rat has emphasized the strong triggering of REM sleep by glutamatergic excitation of this region.64,91 However, glutamatergic excitation of this region in the cat also increases REM sleep,92 suggesting that the difference in response in the two species does not indicate a fundamental difference in control features, although it does indicate species differences in the relative potency of these transmitters or perhaps in the pattern of distribution of receptors for them.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


