Chapter 23 Restless legs and peripheral movement disorders
Restless legs syndrome and periodic movements of sleep
The term “restless legs” has been applied to a number of conditions. Ekbom (1960) originally applied this term to unpleasant crawling sensations in the legs, particularly when sitting and relaxing in the evening, which disappeared on walking. The syndrome was probably first described by Thomas Willis in 1685. “Restlessness” is also a characteristic feature of akathisia, but here the feeling is of inner restlessness not specifically referred to the legs, although this inner feeling can be dissipated by activity. “Inner tension” is also a feature of the urge preceding tics, relieved by the involuntary movement.
The restless legs syndrome (RLS) is characterized by a deep, ill-defined discomfort or dysesthesiae in the legs, which arises during prolonged rest, or when the patient is drowsy and trying to fall asleep, especially at night (Winkelmann et al., 2000; Bassetti et al., 2001, Trenkwalder et al., 2009; Trenkwalder and Paulus, 2010). The disorder is truly diurnal; the symptoms are worse during the night, even when the person tries to stay awake for long periods of time (Hening et al., 1999; Trenkwalder et al., 1999). The discomfort may be difficult to describe – terms such as crawling, creeping, pulling, itching, drawing, or stretching are used, and the feeling usually is felt in the muscles or bones. These intolerable sensations are relieved by movement of the legs or by walking. The feeling usually is bilateral and the arms are rarely involved. Standardized criteria have been put forward by the International Restless Legs Syndrome Study Group (Table 23.1) (Allen et al., 2003). Complaints of restless legs are common, with an estimated prevalence of 3–10% (Phillips et al., 2000; Rothdach et al., 2000; Hening et al., 2004; Bjorvatn et al., 2005). A large population study (over 16 000 adults) showed a prevalence of any restless symptoms to be 7.2%, and moderately or severely distressing symptoms to be 2.7% (Allen et al., 2005). RLS is generally a condition of middle to old age, but at least one-third of patients experience their first symptoms before the age of 20 years (Kotagal and Silber, 2004). Most patients have mild symptoms to begin with, but these worsen with time, so that they seek aid in middle life. Remission is uncommon, occurring in about 15% (Walters et al., 1996). RLS can even affect a phantom limb (Skidmore et al., 2009; Vetrugno et al., 2010).
Table 23.1 Restless legs syndrome
| Essential diagnostic criteria for restless legs syndrome |
1 An urge to move the legs, usually accompanied or caused by uncomfortable and unpleasant sensations in the legs. (Sometimes the urge to move is present with the uncomfortable sensations and sometimes the arms or other body parts are involved in addition to the legs.) 2 The urge to move or unpleasant sensations begin or worsen during periods of rest or inactivity such as lying or sitting. 3 The urge to move or unpleasant sensations are partially or totally relieved by movement, such as walking or stretching, at least as long as the activity continues. |
| Supportive clinical features of restless legs syndrome |
| Family history |
| The prevalence of RLS among first-degree relatives of people with RLS is 3–5 times greater than in people without RLS. |
| Response to dopaminergic therapy |
| Nearly all people with RLS show at least an initial response to either levodopa or a dopamine-receptor agonist at doses considered to be very low in relation to the traditional doses of these medications used for the treatment of Parkinson disease. This initial response is not, however, universally maintained. |
| Periodic limb movements (during wakefulness or sleep) |
| Periodic limb movements in sleep (PLMS) occur in at least 85% of people with RLS; however, PLMS also commonly occur in other disorders and in elderly people. In children, PLMS are much less common than in adults. |
From Allen RP, Picchietti D, Hening WA, et al. Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med 2003;4(2):101–19, with permission.
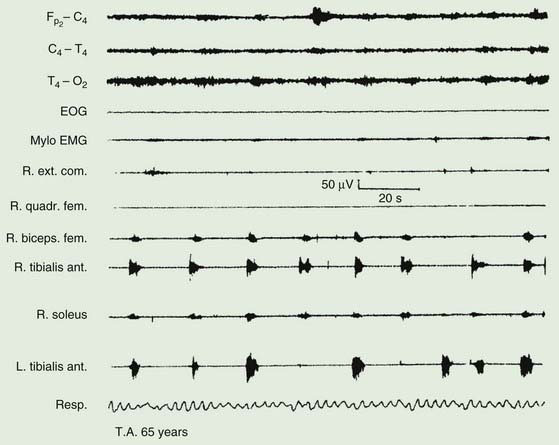
The majority of those with RLS also exhibit periodic movements of sleep (Walters, 1995; Trenkwalder et al., 1996) (Table 23.2). These consist of brief (1–2 seconds) jerks of one or both legs, consisting of, at it simplest, dorsiflexion of the big toe and foot. Initially there is a jerk, but subsequently there is sustained tonic spasm. Such events tend to occur in runs every 20 seconds or so for minutes or hours. Sometimes the whole leg or both legs may flex (Fig. 23.1). The movement resembles a flexion reflex (Bara-Jimenez et al., 2000). Such periodic movements often wake the sleeping partner and may cause disturbance of sleep in the affected individual, in which case there may be excessive daytime drowsiness. Generally they appear during periods of arousal during sleep in stage I and II, and decrease during deep sleep during stages III and IV; they are unusual during REM sleep. Sometimes such flexion movements of one or both legs can occur in the waking subject, particularly when drowsy (Hening et al., 1986) (Video 23.1). Note should be made that some patients with RLS have propriospinal myoclonus just before falling asleep (Vetrugno et al., 2005). 
Table 23.2 Periodic movements of sleep
There is evidence to suggest that the disorder in many if not most patients is transmitted as an autosomal dominant trait (Walters et al., 1996; Winkelmann et al., 2000; Xiong et al., 2010). For a number of years, family studies have been conducted looking for genes with strong mendelian influence. Linkage on 12q seemed the best defined and has been designated RLS1 (Desautels et al., 2005). Variants in the neuronal nitric oxide synthase gene (NOS1) may be the relevant gene at this locus (Winkelmann et al., 2008). Linkage has been found on 14q for a few families and has been designated RLS2 (Bonati et al., 2003; Levchenko et al., 2004). Linkage on 9p24–22 has been designated RLS3 (Liebetanz et al., 2006; Winkelmann and Ferini-Strambi, 2006). Subsequent linkages have identified on chromosomes 2q, 20p, and 6p and designated RLS4, RLS5, and RLS6 (Kemlink et al., 2008). Specific genes, however, have not been identified.
Association studies in large populations have now identified several common sequence variants (single nucleotide polymorphism, SNP) that convey substantial risk for the disorder (Winkelmann, 2008). These variants explain a considerable amount of the familial incidence, and to some extent explain why the earlier studies were having difficulty, since they did not take these strong genetic effects into account. One SNP was identified by two groups, BTBD9 (Stefansson et al., 2007; Winkelmann et al., 2007). Carriers have a 50% risk of developing RLS. It is possible that this SNP is actually specific for periodic limb movements in sleep (PLMS) rather than RLS (Stefansson et al., 2007). Four other SNPs have been identified, MEIS1, MAP2K5, LBXCOR1, and PTPRD (protein tyrosine phosphatase receptor type delta) (Winkelmann et al., 2007; Schormair et al., 2008). The first three of these SNPs have been confirmed in a large replication study (Kemlink et al., 2009). Their biologic function is not clear.
Some cases have been associated with anemia, pregnancy, chronic myelopathies and peripheral neuropathies, gastric surgery, uremia, and chronic lung disease (Ondo and Jankovic, 1996; Winkelmann et al., 2000). It is not uncommon in Parkinson disease (Ondo et al., 2002) and one epidemiologic study found increased prevalence compared with the general population (Krishnan et al., 2003) while another did not (Tan et al., 2002). A point of confusion in this regard is that wearing-off phenomena may mimic RLS symptoms (Peralta et al., 2009). These symptomatic cases of restless legs should be distinguished from the primary familial form of the condition. Occasionally drugs (neuroleptics and antidepressants, lithium, and anticonvulsants) may precipitate intense restlessness of the legs.
Interestingly, in the idiopathic form of the disorder, Earley and colleagues (2000b) found low CSF ferritin levels and high CSF transferrin levels. There was no difference, however, in serum ferritin and transferrin levels. The findings suggest that there might be low brain iron in these patients. Further investigations by this group have shown that the CSF ferritin is low only in the early-onset RLS patients and that levels are lower at night than during the day (Earley et al., 2005). Neuroimaging studies of iron and a neuropathologic evaluation of seven brains has demonstrated decreased iron and H-ferritin in the substantia nigra (Connor et al., 2003). There was a positive correlation between the serum and CSF ferritin levels in both patients with RLS and normal controls, but the slope of the regression lines for the RLS group was lower. These results indicate low brain iron concentration might be caused by the dysfunction of iron transport from serum to central nervous system (CNS) in patients with idiopathic RLS (Mizuno et al., 2005), and now there is good evidence for this (Connor et al., 2011). Another observation is that the number of mitochondria and the mitochondrial ferritin is increased in the substantia nigra; the authors suggest that the mitochondria might also be partially responsible for the low cytosolic iron (Snyder et al., 2009). Iron deficiency could well influence dopamine metabolism (Connor, 2008; Connor et al., 2009).
The pathophysiology of primary restless legs and periodic movements of sleep is unknown (Hening, 2004; Trenkwalder and Paulus, 2004). That dopaminergic mechanisms are involved is strongly suggested by the amelioration of symptoms with dopaminergic therapy. A critical role for the basal ganglia is suggested by the observation that pallidotomy or deep brain stimulation of the pallidum for Parkinson disease ameliorated the sensory symptoms of restless legs (Rye and DeLong, 1999; Okun et al., 2005). (In relation to surgery for Parkinson disease, some patients with deep brain stimulation of the subthalamic nucleus will develop RLS (Kedia et al., 2004). This might be due to the fact that dopaminergic drug therapy is reduced, and this might unmask the disorder.) There is some evidence for D2 receptor binding in the striatum to be low, while presynaptic dopamine function appears normal as indicated by dopamine transporter measurement (Michaud et al., 2002). All studies do not find this D2 receptor abnormality (Eisensehr et al., 2001). Interestingly, there is a strong relationship between iron and dopamine, iron deficiency causing a dopamine deficiency (Allen, 2004).
Voxel-based morphometry (VBM) studies in RLS have produced conflicting results. Etgen et al. (2005) found bilateral increase in gray matter in the pulvinar of the thalamus bilaterally. Unrath et al. (2007) found a decrease in cortical gray matter in the sensorimotor cortex, and the degree of abnormality appeared to correlate with the severity of the disorder. This has some interest since the primary symptom of RLS is sensory. On the other hand, other reports found no apparent relevant abnormality (Hornyak et al., 2008; Celle et al., 2010).
Opioid receptor availability evaluated with positron emission tomography and [11C]diprenorphine, a nonselective opioid receptor radioligand, showed no difference between patients and controls (von Spiczak et al., 2005). However, patients’ symptoms were inversely proportional to the binding in the brain medial pain system.
RLS is characterized by abnormal sensations, and sensory testing reveals abnormalities of temperature sensation. Studies suggest that in idiopathic RLS, the abnormality is in the central processing (Schattschneider et al., 2004; Tyvaert et al., 2009).
If the syndrome is distressing, drug treatment may be justified (Satija and Ondo, 2008). A nocturnal dose of a levodopa preparation is beneficial (Tan and Ondo, 2000; Trenkwalder et al., 2003). Dopamine agonists are preferred such as bromocriptine (Earley et al., 1998; Pieta et al., 1998), pergolide (Wetter et al., 1999; Stiasny et al., 2001), pramipexole (Montplaisir et al., 1999; Ferini-Strambi et al., 2008; Inoue et al., 2010), and ropinirole (Adler et al., 2004; Bogan et al., 2006; Hansen et al., 2009). Dopaminergic therapy with the rotigotine patch can be effective (Trenkwalder et al., 2008a). Dopaminergic therapy has efficacy even in patients with complete spinal cord lesions, suggesting some action at the level of the spinal cord (de Mello et al., 1999). Alternatively, a nocturnal dose of an opiate such as codeine phosphate (Becker et al., 1993; Walters et al., 1993; Prinz, 1995), or of a benzodiazepine such as clonazepam may be of help. Carbamazepine also may help (Telstad et al., 1984), as may baclofen (Guilleminault and Flagg, 1984), clonidine (Wagner et al., 1996), gabapentin (Garcia-Borreguero et al., 2002; Albanese and Filippini, 2003), and pregabalin (Allen et al., 2010; Garcia-Borreguero et al., 2010). It may be necessary to change from one drug to another if tolerance develops. Placebo responsiveness in RLS is very high and this must be taken into account when analyzing results from studies (Fulda and Wetter, 2008).
A problem with dopaminergic therapy of RLS is augmentation, an increase in the severity of symptoms, a shift in time for the start of symptoms to earlier in the day, a shorter latency to symptoms when resting, and sometimes spread of symptoms to other body parts (Hogl et al., 2010). The explanation is not completely clear, but one hypothesis is that there is an increase of dopamine concentration in the CNS with dopaminergic overstimulation, particularly of the D1 receptor (Paulus and Trenkwalder, 2006). This suggests that dopaminergic therapeutic levels should be low for optimal therapy (Williams and Garcia-Borreguero, 2009). It is possible to give the dopaminergic therapy earlier in the day to combat this, but this may provoke an even earlier onset of symptoms. Iron supplementation and opiates have also been suggested for therapy.
Pathologic gambling and other compulsive behaviors have been reported in RLS patients on dopamine agonists, similar to that seen in Parkinson disease (Driver-Dunckley et al., 2007; Quickfall and Suchowersky, 2007; Tippmann-Peikert et al., 2007; Pourcher et al., 2010).
Iron also has been recommended for therapy (Ekbom, 1960), and it has been suggested that it may act by virtue of its effect upon dopamine and opiate receptors (Earley et al., 2000a), but one study was negative (Davis et al., 2000). Another open-label study evaluated the effects of a single 1000 mg intravenous infusion of iron dextran (Earley et al., 2004). Therapy significantly improved the mean global RLS symptom severity, total sleep time, hours with RLS symptoms and PLMS, but on an individual basis failed to produce any response in 3 of the 10 subjects who were fully treated. An open-label study of 25 severely refractory patients with iron dextran produced a good response in some, but the effect was highly variable, and two patients had an anaphylactic reaction (Ondo, 2010). A randomized, double-blind, placebo-controlled study of intravenous iron sucrose did not show any efficacy (Earley et al., 2009). Iron should be curative, of course, in those cases associated with iron deficiency. In patients with low ferritin levels, oral ferrous sulfate 325 mg twice daily improved symptoms more than placebo (Wang et al., 2009). Patients with mild to moderate iron deficiency treated with intravenous iron sucrose also improved, but not significantly more than placebo treatment (Grote et al., 2009).
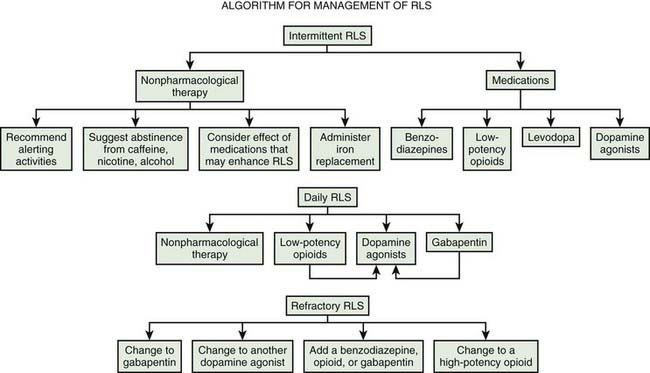
Practice parameters for treatment have been published by the American Academy of Sleep Medicine (Littner et al., 2004). Evidence-based reviews have been published by the European Federation of Neurological Societies (Vignatelli et al., 2006), the Movement Disorder Society (Trenkwalder et al., 2008b), and Cochrane (Scholz, 2011a, 2011b). A review of the dopamine agonists shows their efficacy (Zintzaras et al., 2010). A useful general algorithm has been developed (Fig. 23.2) (Silber et al., 2004).
Peripheral movement disorders
Abnormal involuntary movements (dyskinesias) usually are caused by brain damage or dysfunction. Occasionally, however, lesions of the spinal cord, spinal roots, cervical or lumbar plexus, or even peripheral nerves appear to cause a variety of dyskinesias (Table 23.3). Sometimes the relationship between the trauma and the movement disorder is not definite, and there are no proven rules to relate them. Jankovic and colleagues have proposed some criteria that can be used as guidelines while waiting for more definitive rules (Table 23.4) (Jankovic, 1994; Cardoso and Jankovic, 1995). An example of a definitive peripheral disorder is hemifacial spasm, where compression of the facial nerve by a cerebellopontine angle mass lesion, or by aberrant arteries in the posterior fossa, produces repetitive clonic and tonic contractions of one side of the face. Local pathology in the spinal cord may lead to focal spinal segmental myoclonus. Similar focal myoclonus is sometimes due to damage to spinal roots, the plexus, or peripheral nerves. Such lesions also rarely cause other dyskinesias, such as dystonia and other forms of muscle spasms, sometimes associated with causalgia and reflex sympathetic dystrophy. Finally, a peripheral injury may act as the trigger to the appearance of dyskinesias thought to arise in the brain, as is the case in a significant proportion of patients with primary dystonia. In some way, the peripheral injury alters CNS activity to generate involuntary movements.
Table 23.3 Peripheral movement disorders
Table 23.4 Criteria for a movement disorder to be related to trauma
From Jankovic J. Post-traumatic movement disorders: central and peripheral mechanisms. Neurology 1994;44:2006–14; and Cardoso F, Jankovic J. Peripherally induced tremor and parkinsonism. Arch Neurol 1995;52:263–70.
Hemifacial spasm
Hemifacial spasm is characterized by synchronous spasms of one side of the face (Table 23.5). Most cases are primary, but some are secondary following recovery from facial nerve paresis (Colosimo et al., 2006; Yaltho and Jankovic, 2011). The spasms are usually very brief, but can occur in runs and are occasionally tonic. The disorder typically begins around the eye and this often is the most symptomatic aspect to the disorder (Fig. 23.3 and Video 23.2). The disorder can be bilateral, but then the two sides of the face do not spasm in synchrony. Cases seem to be more common in persons of Asian origin (Poungvarin et al., 1995). Twitching can be brought out by facial muscle contraction. The disorder clearly involves the facial nerve, and the etiology appears to be most frequently (94%) a compression of the nerve by a blood vessel just as the nerve leaves the brainstem (Tan and Chan, 2004; Naraghi et al., 2007). About 4% of cases are due to a tumor compressing the nerve (Han et al., 2010). Biopsy of the compressed nerve shows demyelination. Definitive treatment can be by surgery to decompress the nerve (Samii et al., 2002; Huh et al., 2008; Hyun et al., 2010), although many patients prefer botulinum toxin treatment, which can be highly effective (Poungvarin et al., 1995; Jost and Kohl, 2001; Defazio et al., 2002; Simpson et al., 2008; Bentivoglio et al., 2009; Gill and Kraft, 2010) and can improve quality of life (Tan et al., 2004). 

Nerve origin hypothesis
This hypothesis proposes that the abnormal discharges producing the spasms come from the region of demyelinated nerve under the compression (Nielsen, 1984a, 1984b; Nielsen and Jannetta, 1984). It is known that demyelinated nerve can produce spontaneous discharges, called ectopic discharges. In addition, there can be lateral transmission of activity between demyelinated nerve axons, called ephaptic transmission. Ephaptic transmission can be responsible for involvement of much of the face. It is also possible in the case of activity in demyelinated axons with ephaptic transmission for trains of activity to be produced following a single action potential. These phenomena could explain many of the clinical features.
Additionally, there are physiological studies that are consistent. If a branch of the seventh nerve is stimulated, in these patients there will be late responses seen in muscles innervated by other branches at latencies consistent with ephaptic transmission at the site of demyelination. This phenomenon is not influenced by botulinum toxin treatment (Geller et al., 1989). Studies of the variability of transmission of this effect, using the technique of jitter, are consistent with only the neuromuscular junction and no intervening synapses (Sanders, 1989).
Facial nucleus hypothesis
According to this theory, the peripheral lesion leads to hyperexcitability of the facial nucleus and the discharges arise there. There is a rat model where such a phenomenon has been demonstrated. Perhaps the most persuasive argument for this hypothesis is that there is hyperexcitability of the blink reflex in hemifacial spasm, and this must involve brainstem synaptic circuitry. By this theory, the late responses seen with stimulation of branches of the nerve are enhanced F-waves (Roth et al., 1990; Ishikawa et al., 1994). Lastly, while the calculations deal in differences of only a millisecond or two, conduction times may be more consistent with transmission all the way to the brainstem and back, rather than just to the site of demyelination (Moller and Jannetta, 1984; Moller, 1987).
Focal myoclonus due to root, plexus, or peripheral nerve lesions
Myoclonic jerking of the paraspinal muscles due to a malignant tumor involving the fifth thoracic root, without long tract signs of spinal cord involvement, has been described (Sotaniemi, 1985). Similar focal myoclonus of the legs has also occurred with lumbosacral radiculopathy, and after lumbar laminectomy for lumbar stenosis and root lesions (Jankovic and Pardo, 1986). Rhythmic myoclonus of the quadriceps muscle has been reported due to a Schwann-cell sarcoma of the femoral nerve (Said and Bathien, 1977).
Focal myoclonus of the right arm due to a brachial plexus lesion has been described following radiotherapy for carcinoma of the breast followed by abduction trauma of the right shoulder (Banks et al., 1985). The latter case exhibited rhythmic muscle jerks at about five per second in the distribution of the axillary and radial nerves, but not in other muscles innervated by the lateral and medial cords of the brachial plexus. Electromyographic analysis of this case indicated that the myoclonus arose from a generator located in a segment of the posterior cord of the brachial plexus, between the departure of the axillary nerve and distal to the emergence of the suprascapular nerve. Another patient developed myoclonus of one arm after an electrical injury to the left brachial plexus (Jankovic and Pardo, 1986). Myoclonus of an arm has even occurred after a thoracic sympathectomy (Jankovic and Pardo, 1986).
Swanson et al. (1962) suggested two mechanisms which might be responsible for spinal segmental myoclonus: (1) enhanced neuronal excitability due to direct cellular excitation by inflammation or tumor, or (2) enhanced neuronal excitability due to removal of inhibition. The former seems unlikely, since spinal segmental myoclonus can occur without evidence of damage to anterior horn cells. Loss of inhibition of anterior horn cell pools seems more probable.
Posterior rhizotomy or hemicordectomy leads to abnormal spontaneous discharge of some spinal neurons in the deafferented segments, which tend to fire in bursts at high frequency (Loeser and Ward, 1967). However, these bursting spinal neurons are found in the dorsal, not the ventral horns. Nevertheless, such spontaneous bursting of spinal interneurons following deafferentation might drive anterior horn cells to produce focal myoclonus.
Alternatively, loss of inhibitory spinal interneurons might liberate anterior horn cells to fire spontaneously in a rhythmic burst fashion. In the case described by Davis et al. (1981) spinal myoclonus occurred following ischemic damage to the cord, which at autopsy was found to have caused extensive loss of small and medium-sized interneurons, with relative preservation of large anterior horn cells. The loss of inhibitory spinal interneurons could release anterior horn cells to discharge spontaneously, but what then determines their tendency to fire repetitively and rhythmically is less clear (Kiehn, 1991; Kiehn and Eken, 1997). Loss of spinal interneurons also is the pathologic change thought to be responsible for alpha spinal rigidity.
Jumpy stumps
Not only did Weir Mitchell (Mitchell, 1872) describe causalgia after gunshot wounds of peripheral nerves, he also recorded tremor, jerks, and spasms of the remaining stump following amputation, sometimes associated with severe phantom pain. The “painful, jumpy stump” has since been described by others (Russell, 1970; Steiner et al., 1974; Marion et al., 1989; Kulisevsky et al., 1992), and even a phantom dyskinesia induced by metoclopramide has been recorded (Jankovic and Glass, 1985) (Video 23.3). 
Jerking of the amputation stump (jactitation), coinciding with lancinating neuralgic stump pains, frequently occurs in the postoperative period but settles over weeks or months (Russell, 1970). The cases referred to above, however, experienced spasms and jerks of the stump for prolonged periods, for example up to 40 years in one of the patients reported by Marion et al. (1989), who also reviewed many similar cases described in the earlier literature. Jerking of the stump frequently was preceded by severe pain in the stump, appearing weeks or months after the surgery. Upper or lower limb stumps could be affected. The stump jerks could be induced by voluntary movement or, sometimes, by cutaneous stimuli.
Steiner et al. (1974) considered involuntary stump movements to be a form of segmental myoclonus, caused by afferent impulses arising from the severed nerves. Marion et al. (1989) concluded that they were due to either “the result of functional changes in spinal (or cortical) circuitry leading to redirection of afferent information through different spinal neurons, or structural reorganization of local neuronal circuitry by axonal sprouting following nerve injury.”
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


