Figure 62.1. Rufinamide. MW 238.2, (C10H8F2N4O).
Rufinamide is nearly insoluble in water and slightly soluble in methanol and ethanol. This would make it difficult to prepare an intravenous formulation. Solubility in water and gastric fluid is approximately 40 to 70 mg/L at 37ºC. Dissolution is the rate-limiting step for absorption. Rufinamide forms a white crystalline powder and is compacted into scored film tabs of 100-, 200-, and 400-mg tablets.
MECHANISMS OF ACTION
Rufinamide modulates voltage-dependent neuronal sodium channels; however, it also inhibits seizures triggered by γ-aminobutyric acid (GABA) antagonists, and its anticonvulsant mechanisms in humans are unknown. The drug interacts with sodium channels in cultured rodent cortical neurons, prolongs inactivation of voltage-dependent sodium channels in spinal cord neurons, and acts to reduce repetitive firing of sodium-channel dependent neurons (1). The drug does not interact, however, with several subtypes of rodent and human voltage-gated sodium channels: rat Nav1.2a, rat Nav1.8, and human Nav1.5 (2). Interactions with sodium channel isoforms, such as human Nav1.2, involved in familial epilepsy syndromes have not been evaluated.
Rufinamide’s effects in prolonging inactivation of voltage-dependent sodium channels are consistent with its potent inhibition of maximal electroshock–triggered seizures in rodents (oral ED50 = 4 to 24 mg/kg) (3). Rufinamide’s inhibition of maximal electroshock seizures (MES) was additive with other AEDs, but it did not potentiate or reduce the effects of other AEDs (4). Rufinamide also prevents clonic seizures induced by injected (s.c. and i.p.) pentylenetetrazole (PTZ) in mice, but did not prevent seizures caused by oral PTZ treatment. Rufinamide caused behavioral toxicity on the rotarod test only at extremely high doses; consequently, rufinamide’s protective indexes for MES and PTZ tests are much higher than traditional AEDs (e.g., phenytoin in MES model, valproic acid in PTZ model) (3).
Rufinamide inhibits seizures induced by the GABA-A antagonists bicuculline and picrotoxin, with less effect on strychnine-induced seizures. Rufinamide does not inhibit seizures in the WAG/Rij rat, however, which is a genetic model of absence epilepsy with GABA-A receptor abnormalities (5). Rufinamide also does not interact directly with GABA receptors or modulators. This suggests rufinamide’s influences on cortical inhibition are indirect, possibly mediated by modulation of voltage-dependent sodium channels in cortical interneurons.
Rufinamide has mixed effects on chronic seizure models: it delayed the development of electrically kindled afterdischarges in the cat, but not in the rat. It markedly reduced recurring motor seizures induced by aluminum hydroxide placed on monkey cortex (6). Overall, these studies suggest that rufinamide modulation of voltage-dependent sodium channels may indirectly influence seizures via effects on cortical inhibition. However, associations between these mechanisms and effects on seizures associated with Lennox–Gastaut syndrome are unknown.
ABSORPTION, METABOLISM, AND DRUG INTERACTIONS
The pharmacologic profile for rufinamide is summarized in Table 62.1.
Table 62.1 Pharmacokinetic Properties of Rufinamide

Rufinamide is well absorbed orally in the fed state (≥85% absorption in healthy volunteers) with a slow rate of absorption; absorption decreases slightly at high doses (7). The relative extent of absorption of rufinamide was lower at a dose of 1600 mg/day compared to 200 to 800 mg/day in a large pharmacokinetic study (8). Bioavailability of single doses of rufinamide is increased by food; however, food effects were not seen with chronic dosing (9). Patients received rufinamide only with food in clinical trials, and it is approved to be dosed with food. Peak rufinamide concentrations (Tmax) occur approximately 6 hours after dosing when taken with food and approximately 8 hours when dosed while fasting (10). Rufinamide has relatively low (34%) protein binding—mostly to albumin—and is distributed in the bloodstream equally between erythrocytes and plasma (7). Rufinamide’s apparent volume of distribution (Vd) is approximately 50 L at a 3200 mg/day dose and increases slightly with very high doses and high body surface area (11).
Rufinamide is eliminated via hydrolysis into an inactive carboxylic acid metabolite (CGP 47292), which is renally excreted. Less than 2% of rufinamide is recovered in the urine (4). A small fraction of metabolite is glucuronidated and subsequently excreted. Rufinamide is hydrolyzed by a carboxylesterase, which is concentrated in the liver, but is present in brain and other tissues (7). Rufinamide does not induce carboxylesterase and its metabolism is not dependent on cytochromes; thus, major drug–drug interactions are unlikely (7). With the exception of a valproic acid interaction in children, the overall pharmacokinetics for rufinamide are similar in children and in adults, including the elderly, with clearance proportionate to dose and body surface area (12). However, in one study in children, apparent clearance of rufinamide was higher in younger children than older children even when concomitant valproic acid was taken into account. Similar to that of other studies, concomitant enzyme inducing medications increased clearance in all age groups, particularly so in younger children (13). Moreover, human carboxylesterase 1, which is mainly involved in metabolism of rufinamide, has been shown to be inhibited by valproate or its metabolite valproyl-CoA in vitro, and thus, patients on concomitant valproate may need lower doses of rufinamide (14).
While no studies formally examined the relationship between serum concentrations and clinical response/efficacy of rufinamide, in one pediatric study of patients with epileptic encephalopathies, rufinamide plasma concentrations were extremely variable and no correlation between serum concentration and clinical response could be demonstrated (15). One study reported that plasma concentrations above 15 mg/mL are “useful” although the study found no validated therapeutic range (16).
In clinical studies, rufinamide had only small effects on concentrations of several other AEDs (17): Phenytoin clearance was decreased slightly, with plasma levels increasing 7% to 21%; carbamazepine, lamotrigine and phenobarbital concentrations decreased 7% to 13%; and topiramate and valproate concentrations were unchanged. Patients taking valproic acid, especially children, had increases in rufinamide concentrations (18). Valproic acid caused average increase in rufinamide concentrations of 40% in children and 11% in adults (12). Small children (<30 kg) with very high valproic acid concentrations (e.g., 100 mg/L) had increases in rufinamide concentrations of up to 70%; however, this varied widely across patients (18). Rufinamide doses were not adjusted in clinical trials for patients receiving valproic acid; however, reduced dose reductions of 50% to 60% have been recommended for small children (<30 kg) taking valproic acid (14). European regulators have requested an additional pediatric monitoring study to evaluate this interaction further. Adolescents and adults receiving valproic acid had much smaller increases in rufinamide concentrations compared to children: ≤26% increases in adolescents and <16% increases in adults (18). Other AEDs were associated with variable small-to-moderate decreases in rufinamide concentrations: carbamazepine (19% to 26%), phenobarbital (25% to 46%), phenytoin (25% to 46%), and primidone (25% to 46%). Lamotrigine and topiramate did not alter rufinamide concentrations (6). One pediatric study did not show any significant effect of rufinamide on the kinetics of concomitant AEDs (13). Among adolescents and adults however, one study showed that only valproic acid had any significant effect on rufinamide clearance (decrease) with no other concomitant medication showing any effect (19).
Rufinamide had a modest interaction with oral contraceptives: Repeated administration of 1600 mg/day of rufinamide decreased ethinyl estradiol concentrations by 22% and norethindrone by 14%. It is unclear whether higher doses of rufinamide might produce greater hormonal reductions and contraceptive failure (19). Triazolam clearance increased slightly with rufinamide treatment (4). Increased clearance is most likely caused by modest induction of CYP3A4 and is not believed to be clinically relevant (20).
Due to extensive metabolism, rufinamide elimination is not influenced by renal dysfunction, and no specific dosage changes are required for patients with renal impairments (11). No marked difference in rufinamide concentrations was found in patients experiencing severe renal impairment as compared to healthy individuals after a single 400-mg dose (18). During dialysis, area under the curve (AUC) was decreased by approximately 30%. Simulations over 1 week, however, estimate three 3-hour dialysis sessions would decrease total rufinamide exposure (AUC) by approximately 12%. This suggests that no specific dose adjustment of rufinamide is likely to be required for patients with renal failure undergoing hemodialysis (18).
CLINICAL STUDIES
Lennox–Gastaut Syndrome
Rufinamide is approved with an orphan drug indication for adjunctive treatment of Lennox–Gastaut syndrome in the United States and Europe. Patients with Lennox–Gastaut syndrome typically have multiple seizure types along with encephalopathies. Their most characteristic (and serious) seizure types are tonic and tonic–atonic “drop attacks,” which cause sudden falls and injuries. Patients also have varying patterns of atypical absence, myoclonic, atonic, tonic–clonic, and complex motor seizures. Most patients have slow spike-and-wave discharges and generalized slowing on EEG. Effects of rufinamide in treating seizures in patients with Lennox–Gastaut syndrome were evaluated in a randomized, parallel-design, placebo-controlled study of children and adults >4 years (N = 138) (21). Seizures associated with falls (predominantly tonic and tonic–atonic seizures) and total seizures were assessed. Patients were treated with rufinamide 45 mg/kg, up to a maximum of 3200 mg/day, divided twice a day. Patients receiving rufinamide had a 42.5% median reduction in tonic–atonic seizures compared to a 1.4% increase in seizures for patients receiving placebo treatment (Fig. 62.2A). The patients’ total seizure frequency was reduced 32.7% during rufinamide treatment compared to a median of 11.7% for placebo treatment. Seizure responder rates (proportions of patients with >50% seizure reduction) were also significantly higher for patients treated with rufinamide (42.5%) compared to placebo (16.7%) (Fig. 62.2B).
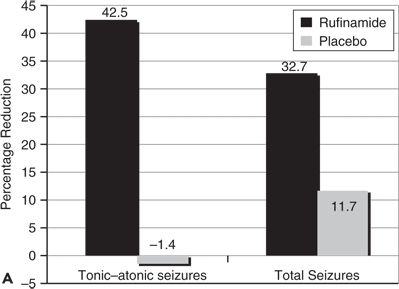
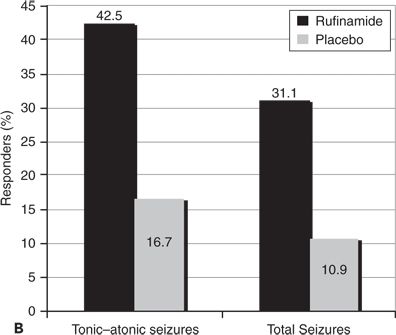
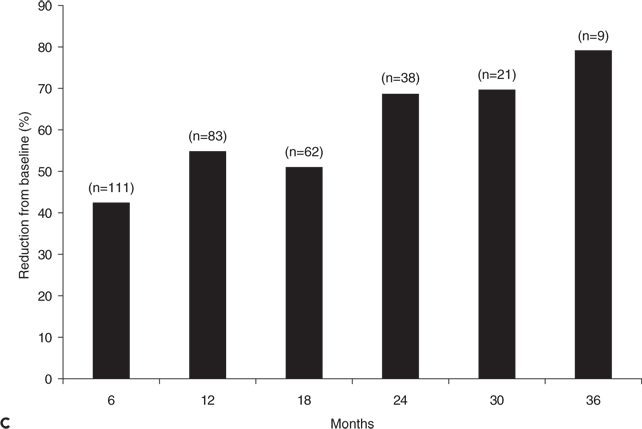
Figure 62.2. A:Median percentage reduction in total seizure frequency and tonic–atonic seizure frequency. B:Percentage of patients (responders) who experienced at least 50% reduction in tonic–atonic seizure frequency. C:Efficacy of adjunctive rufinamide therapy during long-term extension study treatment of patients with Lennox–Gastaut syndrome. (C, Data from Glauser T, Kluger G, Sachdeo R, et al. Open-label extension study of the efficacy and safety of rufinamide adjunctive therapy in patients with Lennox-Gastaut syndrome. Epilepsia. 2005;46(suppl 6):406.)
Efficacy was sustained during open-label extension treatment, with decreases in seizure frequency of 43% to 79% during 6 to 36 months of treatment; patients converting from placebo to rufinamide also had substantial reductions in seizures (Fig. 62.2C) (22). Responder rates for patients, during their most recent 6 months of therapy, were 45.1% for total seizures and 47.9% for tonic–atonic seizures. A total of 9.4% of patients were free of tonic–atonic seizures during their last 6 months of open-label treatment (23).
Most patients with Lennox–Gastaut syndrome tolerated rufinamide well, especially considering that treatment was combined with one to three concomitant AEDs (21) and titrated very rapidly. The most common adverse events (≥10%) seen in rufinamide-treated patients as compared to those receiving placebo were as follows: somnolence (24.3% vs. 12.5%), vomiting (21.6% vs. 6.3%), pyrexia (13.5% vs. 17.2%), and diarrhea (5.4% vs. 10.9%), respectively. Vomiting and somnolence were the only adverse events occurring at an incidence of 5% greater than placebo treatment. Of the 124 patients entering open-label extension treatment (median dose 1800 mg/day), 12 subsequently discontinued treatment due to adverse events (18). The most commonly reported adverse events during the uncontrolled extension treatment phase were vomiting (30.6%), pyrexia (25.8%), upper respiratory tract infection (21.8%), and somnolence (21.0%). Some of these symptoms appeared to be due to childhood viral illnesses rather than due to rufinamide treatment.
Rufinamide’s effectiveness for treating seizures in Lennox–Gastaut syndrome was demonstrated in a single placebo-controlled study, but has generally been confirmed by the long-term extension treatment results and by several open treatment series: 41% of patients were >50% responders in a long-term open-label extension study (24). A multicenter prospective open treatment Italian series reported 51% of children and adults with Lennox–Gastaut syndrome had ≥50% responses to rufinamide; 9.3% were seizure free over a mean follow-up of 12 months of treatment (25). During 12 weeks of maintenance treatment with rufinamide 20 to 40 mg/kg/day in a Korean study, 36% of patients were >50% responders and responders included patients with drop seizures, myoclonic seizures, and “epileptic spasms” (26). Similarly, in a single center Korean study of children and young adults, 35% of patients at 6 months were >50% responders (27).
Small retrospective series have reported some patients with other epileptic encephalopathies may respond to rufinamide treatment (28,29). In 38 patients (ages 17 months to 23 years) with “epileptic spasms,” 53% of patients were >50% responders in one series (30). Other small retrospective series reported benefit in some children with Doose syndrome (myoclonic–astatic epilepsy), malignant migrating partial epilepsy of infancy, and epilepsy with myoclonic absences, with only a small proportion with Dravet syndrome responding to treatment (31).
In a small observational study from Europe, 8 patients with Doose syndrome were treated with rufinamide for 6 to 18 months: 6 out of 8 patients had a >75% reduction in their major seizure type (myoclonic–astatic seizures) immediately after initiation of adjunctive rufinamide; however, this efficacy was not sustained during long-term therapy (32). Another series noted greater rufinamide responses in patients with Lennox–Gastaut syndrome and epileptic spasms (with up to 40% of patients being 50% responders) compared to patients with Dravet syndrome and myoclonic seizures (29).
Partial-Onset Seizure Trials
Rufinamide consistently reduced the frequency of partial-onset seizures in adults in three large randomized, placebo-controlled, multicenter trials. Effect sizes were modest, though, and the drug was not submitted for regulatory approval for this indication. In a large (N = 357) placebo-controlled randomized trial of adults and adolescents (≥16 years) with difficult-to-control partial-onset seizures, rufinamide 3200 mg/day reduced median seizure frequency by 23.2% compared to a 9.8% decrease with placebo treatment (19). In another placebo-controlled randomized trial, rufinamide 3200 mg/day reduced median seizure frequency by 20.4% compared to a 1.6% increase during placebo treatment. Patients not receiving carbamazepine during rufinamide treatment in this study had a slightly higher median seizure reduction of 29% (33). A third large (N = 647) study in adults and adolescent (ages 15 to 65 years) with partial-onset seizures explored treatment with four doses of rufinamide 200, 400, 800, or 1600 mg/day (divided twice a day) and placebo. Patients had a significant linear trend in dose response across the four rufinamide doses (P = 0.003): 50% responder rates ranged from 9% with placebo to 4.7% with rufinamide 200 mg/day, 16% for 400 mg/day (P = 0.027), 12% for 800 mg/day dose (P = 0.012), and 14% with a 1600 mg/day dose (P = 0.016) (Fig. 62.3) (34).
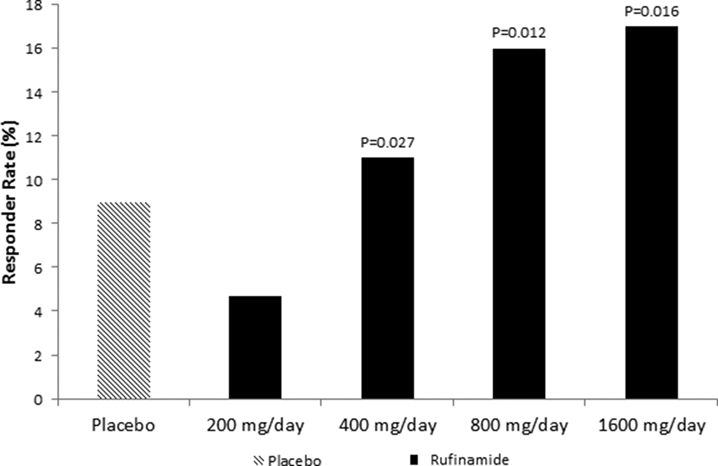
Figure 62.3. Greater than or equal to 50% responder rates for four doses of adjunctive rufinamide therapy compared to placebo treatment among adults and adolescents with partial-onset seizures. (Data from Elger CE, Stefan H, Mann A, et al. A 24-week multicenter, randomized, double-blind, parallel-group, dose-ranging study of rufinamide in adults and adolescents with inadequately controlled partial seizures. Epilepsy Res. 2010;88(2–3):255–263.)
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


