CHAPTER 151 Sarcoidosis, Tuberculosis, and Xanthogranuloma
Sarcoidosis
Sarcoidosis is a chronic, multisystem granulomatous disease of unknown cause. It can occur in any racial or ethnic population and most often develops in persons between the ages of 20 and 40 years. In the United States, its prevalence ranges from 10 to 40 per 100,000, with an approximately 10 : 1 ratio of black to white patients and a slight female preponderance.1,2 Any organ system can be involved in sarcoidosis, with the lungs, lymph nodes, skin, and eyes being the most common. Five percent of patients have neurological manifestations.3–8 Of these patients with neurosarcoidosis (NS), 50% have neurological problems at the time of initial diagnosis. In a third of these patients, more than one neurological manifestation of their disease is already present or will develop.
Histology and Pathophysiology
Although the precise cause of sarcoidosis is not known, there is evidence that the disease results from dysregulation of the cellular immune responses to foreign antigens or self-antigens, thereby leading to granuloma formation.1 These granulomas are composed of epithelioid cells, histiocytes, T cells, monocytes, and fibroblasts. Granuloma formation can ultimately lead to fibrosis and organ dysfunction.
Clinical Features
Cranial Neuropathy
Cranial neuropathy is the most frequently encountered neurological manifestation of sarcoidosis; it occurs in approximately 75% of patients with NS.8 More than 50% of patients have involvement of multiple cranial nerves.
Dysfunction of the facial nerve is the most common cranial neuropathy seen in NS, and it develops in more than 50% of patients.8 The peripheral palsy can be unilateral or bilateral and is often transient.
NS can occur in the subfrontal region and cause an olfactory neuropathy and anosmia. Involvement of the optic nerve is relatively common and is seen in as many as 5% of patients.8 Funduscopic examination can demonstrate optic atrophy, optic disc swelling or papilledema, or retrobulbar optic neuritis.9 In patients with visual or optic abnormalities, however, it must be remembered that sarcoidosis affects the eye as a uveitis far more often than the optic nerve.
Meningitis
Symptoms of meningeal irritation, including headache, nuchal rigidity, nausea, and rarely, fever, have been reported to occur in 3% to 26% of patients with NS.8 Symptomatic meningeal involvement can take the form of either acute aseptic meningitis or chronic meningitis. Cerebrospinal fluid (CSF) studies generally reveal a mild mononuclear pleocytosis with increased protein. Glucose may be normal or decreased, with no evidence of bacteria. Given the extent of meningeal involvement seen in most patients with NS, symptomatic meningitis is relatively rare. Occasionally, meningeal-based granulomatous mass lesions can develop.
Pituitary and Hypothalamic Sarcoidosis
When sarcoidosis involves the CNS parenchyma, it most commonly affects the hypothalamus and parasellar region.10 This can result in vegetative alterations and neuroendocrinologic dysfunction. Symptoms include disturbances in sleep, appetite, thirst, temperature, and libido. Hypothalamic and pituitary lesions can also cause thyroid, gonadal, and adrenal abnormalities. Altered thirst, antidiuretic hormone deficiency or excess, and hyperprolactinemia have been reported.
Space-Occupying Lesions
Granulomatous masses forming localized space-occupying lesions can occur throughout the CNS but are most frequently found in the cerebral hemispheres.6 They can be manifested as a large, solitary mass mimicking either a primary or metastatic brain tumor.11,12 Alternatively, they can appear as multiple nodules.8 Finally, they can take the form of a dural-based plaque-like mass resembling a meningioma.13–16
Seizures
Generalized or partial seizures have been reported in as many as 22% of patients with NS.17 They are thought to be caused by supratentorial parenchymal involvement in which the granulomatous inflammation is often found in a perivascular distribution.
Hydrocephalus
Hydrocephalus secondary to obstruction of normal CSF flow is a relatively common and potentially lethal clinical feature of NS.18–20 Communicating hydrocephalus can result from extensive meningeal involvement causing decreased CSF resorption through the arachnoid villi. Obstructive hydrocephalus can develop secondary to an intracranial mass or inflammation preventing the flow of CSF through the foramen of Monro, aqueduct of Sylvius, or foramen of Luschka or Magendie.21,22
Spinal Cord Sarcoidosis
Sarcoidosis of the spinal cord is relatively rare. It can occur at any segment of the spinal cord, as well as at the cauda equina, and the granulomatous lesions may be intramedullary or extramedullary.23–30
Peripheral Neuropathy
Peripheral neuropathic manifestations include mononeuropathy, mononeuritis multiplex, and generalized sensory, sensorimotor, and motor neuropathies.31,32 Patients can have a small-fiber sensory or autonomic neuropathy.33 Clinical features resembling Guillain-Barré syndrome have also been described. Nerve biopsy shows granuloma formation in both the epineurial and perineurial spaces, as well as axonal degeneration. There can be associated vascular inflammation, thus suggesting that ischemia may play a role in the nerve damage.
Diagnostic and Imaging Studies
The diagnosis of sarcoidosis is firmly established only when the clinical and paraclinical findings are supported by histologic evidence of widespread noncaseating granulomas.1,2 When neurological findings develop in a patient with sarcoidosis, NS should be considered, although an intercurrent infection or malignancy must be excluded. The following diagnostic criteria for NS are adapted from Zajicek and colleagues36:
When NS is suspected, the patient should be evaluated for evidence of systemic disease and a search for a site suitable for biopsy undertaken. Because corticosteroids can eliminate evidence of systemic inflammation, they should be withheld until the diagnostic evaluation is completed unless severe illness requires their use. The search for systemic sarcoidosis should begin with an examination of the skin and lymph nodes.1,2 Chest radiographs are abnormal in approximately 90% of patients with sarcoidosis, but frequently, additional diagnostic information can be obtained from a computed tomography (CT) scan of the thorax. Serum angiotensin-converting enzyme (ACE) levels can be elevated in patients with sarcoidosis. However, infection or malignancy can cause serum ACE levels to be high, and in patients with isolated NS, serum ACE levels may be normal.37,38 Ophthalmologic examination can reveal uveitis or retinal vasculopathy, as well as conjunctival nodules. Endoscopic nasal and sinus examination can show evidence of mucosal inflammation.
CSF abnormalities occur in NS, but they tend to be nonspecific and thus are not a reliable marker of the disease. CSF opening pressure is elevated in approximately 10% of patients, and protein is increased in two thirds of patients. CSF glucose can be normal or low. Approximately 50% of patients have a predominantly mononuclear pleocytosis. The immunoglobulin G index may be elevated, and oligoclonal bands may be present.6,8,39 The concentration of ACE in CSF may be elevated, but this finding is not specific to NS.40 There are reports of patients deteriorating after lumbar puncture.20
Gallium scanning can be informative; uptake in the lungs and salivary, parotid, and lacrimal glands is suggestive of sarcoidosis and, when a distinctive pattern is found in combination with elevated serum ACE levels, may be highly specific (>95%) for sarcoidosis.41 Whole-body fluorodeoxyglucose positron emission tomography (FDG-PET) is a sensitive technique for highlighting areas of inflammation.42
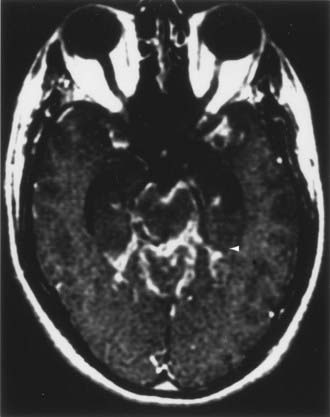
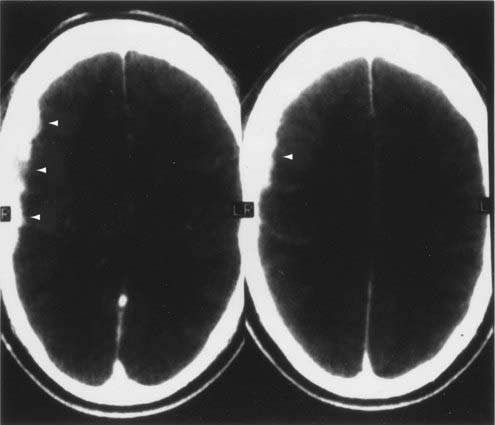
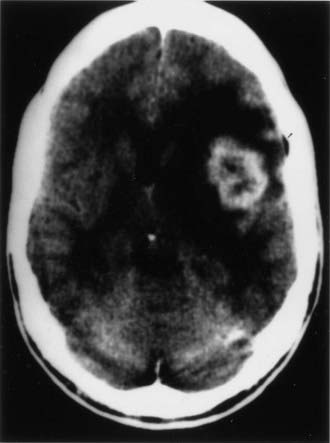
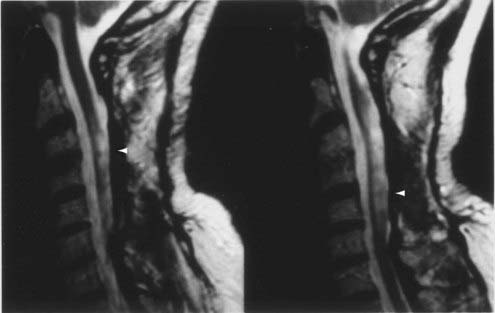
CT and magnetic resonance imaging (MRI) enhance detection of meningeal (Figs. 151-1 and 151-2) and parenchymal disease (Figs. 151-3 and 151-4), even in patients without any neurological findings.
On CT, the granulomatous lesions of NS are usually isodense or hyperdense on non–contrast-enhanced studies and uniformly enhancing after the administration of contrast medium.8,43 CT can reveal bony erosion of the skull base caused by NS lesions. The orbital contents, including the optic nerve and extraocular muscles, can also be visualized, particularly with sagittal and coronal reconstructions.
The imaging procedure of choice for NS is MRI with gadolinium enhancement.44–47 This test is particularly sensitive for detecting disease of the parasellar region, basal cisterns, and spinal cord. On T1-weighted images, NS lesions are usually hyperintense in relation to normal brain parenchyma, and they enhance after the administration of gadolinium. T2-weighted images often show minimal edema surrounding the lesions, although this finding can be variable and may be related to disease activity. A wide spectrum of NS abnormalities can be seen on MRI and can mimic other neurological diseases. Gadolinium-enhanced MRI can reveal both diffuse (see Fig. 151-1) and local (see Fig. 151-2) leptomeningeal involvement.47,48 The dura often shows marked thickening along with evidence of inflammation (pachymeningitis). Parasellar lesions and intraparenchymal masses (see Fig. 151-3) can also be readily identified, but the latter can easily be mistaken for a neoplasm.49 Involvement of the optic nerves or other cranial nerves can likewise be documented. MRI can identify hydrocephalus, and the presence of transependymal edema on T2-weighted images can be a useful indicator of increased intracranial pressure secondary to obstruction of CSF flow. Finally, MRI can be valuable in the detection of sarcoidosis involving the spinal cord (see Fig. 151-4) and cauda equina.50
In addition to its utility in identifying lesions consistent with NS, gadolinium-enhanced MRI can be used to monitor the efficacy of NS treatment.51 Decreased enhancement and regression of the size of lesions suggest therapeutic efficacy.
Biopsy of the brain, spinal cord, meninges, nerve, or muscle is occasionally performed to confirm a diagnosis of NS in patients with known sarcoidosis who have a neurological problem consistent with NS. Biopsy should also be considered in patients with known systemic sarcoidosis and neurological disease who are progressively deteriorating despite “optimal” treatment to uncover an alternative diagnosis.52 Importantly, biopsy can support a diagnosis of NS in patients in whom the diagnosis is suspected but for which there is no confirmatory systemic documentation. Occasionally, NS is the unexpected consequence of a CNS biopsy, for instance, in a patient with a dural-based mass thought to be a meningioma but on biopsy granulomatous inflammation is found.
Treatment
Because of the relative rarity of NS, there have been no rigorous clinical trials to determine optimal treatment. Given the pathophysiology of sarcoidosis, however, immunosuppressive therapy has been the cornerstone of therapy.6,8
Corticosteroids are the first-line agents for the treatment of NS. The site of neurological involvement, disease severity, and response to treatment generally dictate the dose and duration of corticosteroid therapy. For example, patients with facial nerve palsy or aseptic meningitis are often treated with prednisone, 0.5 mg/kg per day for 2 weeks; patients with myopathy or neuropathy are generally treated with prednisone, 0.5 mg/kg per day for 4 weeks, and then reassessed, with most patients requiring chronic therapy; and patients with a meningeal or parenchymal mass lesion, encephalopathy, or symptomatic hydrocephalus usually require prednisone, 0.5 to 1 mg/kg per day for 4 weeks, before any improvement is appreciated, and most require chronic therapy. Once the acute exacerbation is under control, corticosteroid administration can usually be slowly tapered. Frequent clinical evaluation and, for CNS disease, periodic MRI to assess the extent of enhancement can be helpful in guiding therapy.51,53
Patients who deteriorate despite aggressive corticosteroid therapy, who cannot tolerate corticosteroids, or who have a contraindication to corticosteroids may benefit from alternative therapeutic agents.54–56 Mycophenolate mofetil, cyclosporine,57 azathioprine, methotrexate,58,59 chlorambucil, cyclophosphamide,55 hydroxychloroquine,60 pentoxifylline,61,62 thalidomide,63,64 and infliximab and adalimumab65–68 have all been used to treat NS. There are no studies comparing the efficacy of these alternative treatments in patients with NS. Consequently, selection of agents is often based on ease of use, cost, and desire to avoid the complications of a particular drug. Because a patient’s response to a specific agent cannot be predicted, two or three different agents should be tried before concluding that the patient’s disease is refractory.
Cranial or spinal irradiation has been used for refractory NS and should be considered in patients who fail corticosteroid therapy and trials of alternative agents.69,70 Radiotherapy has also been used in patients with acute, life-threatening disease.
Intracranial and intraspinal mass lesions generally respond well to corticosteroids or alternative agents if the disease has been caught early, before the development of significant ischemia or fibrosis.6,8 Thus, in patients with known systemic sarcoidosis in whom a mass lesion develops, corticosteroid therapy should be initiated promptly as long as there are no significant contraindications and infection and malignancy have been reasonably excluded as diagnoses. In patients without extensive systemic disease, treatment of an intracranial or intraspinal mass can be complicated by the imaging similarities of sarcoidosis, tuberculosis, fungal disease, and neoplasm.43 In patients without known sarcoidosis, it may be necessary to perform a biopsy at the primary site of involvement to determine the nature of a mass lesion. During surgery, effort should be made to minimize disruption of the surrounding parenchyma because NS can be infiltrative and patients can deteriorate after surgical intervention. If the biopsy reveals noncaseating granulomas, further resection should be deferred while appropriate medical management is pursued. Only in highly refractory cases in which a mass lesion persists or enlarges despite optimal immunosuppressive therapy should surgical debulking or resection be considered. The results of these aggressive surgical interventions are often unsatisfactory.71
Patients with chronic, asymptomatic hydrocephalus can be observed clinically, and surgical intervention can be offered if the hydrocephalus worsens or if the patient deteriorates despite medical therapy. These patients must be watched closely, however, because abrupt, life-threatening deterioration can occur. In patients who respond to medical therapy, improvement in CSF flow can occur over time.72
Conclusion
The long-term course of NS has not been clearly defined. Approximately two thirds of patients have a monophasic illness, and the others have either a relapsing-remitting course or progressive disease.72 As many as 10% of NS patients die as a result of the inflammatory disease process or its treatment. These patients typically have CNS parenchymal disease or hydrocephalus. Prompt diagnosis of NS can lead to earlier treatment, and this may improve the outcome.
Tuberculosis
Tuberculosis (TB), a worldwide disease, experienced a resurgence in the United States in the early 1990s, largely related to the human immunodeficiency virus (HIV) epidemic, immigration from countries endemic for TB, transmission of TB in settings such as hospitals and prisons, and the development of multidrug-resistant strains of TB. TB is primarily a disease of the lungs, but extrapulmonary involvement is common. CNS involvement is reported in as many as 10% of immunocompetent patients with TB.73 CNS TB is a defining condition of acquired immunodeficiency syndrome (AIDS), and both TB and CNS involvement are more prevalent in the HIV-infected population.74 CNS involvement can take the form of tuberculous meningitis, intracranial and spinal cord tuberculomas, abscesses, and Pott’s disease. Here, the focus is primarily on neurosurgical management of CNS tuberculomas and abscesses.
Pathogenesis
CNS TB is almost entirely due to infection with Mycobacterium tuberculosis. The bacterium is transmitted by inhalation of aerosolized droplets, and as few as 1 to 10 organisms are sufficient to cause infection. As the bacteria multiply in alveoli and macrophages, a cell-mediated immune response ensues and generates a granulomatous reaction characterized by a tubercle and caseating necrosis. If the infection is not contained, bacteria can spread hematogenously to distant sites such as the CNS. The inciting CNS tubercle is called a Rich focus.75 The location of tuberculous lesions in the brain is related to the pattern of blood flow, and lesions generally involve the corticomedullary junction and periventricular regions. They usually occur in the cerebral hemispheres and basal ganglia in adults and in the cerebellar hemispheres in children; brainstem and spinal lesions are rare.76 Tubercles that rupture into the subarachnoid space cause tuberculous meningitis, whereas deep-seated tubercles cause tuberculomas or abscesses. The immune reaction around a tuberculous focus can also cause vascular inflammation, vasculitis, and edema. Vasculitis can lead to ischemia, as well as poor delivery of drug to affected areas. Furthermore, hydrocephalus is common in CNS TB, especially in children.
Tuberculomas are usually solitary lesions.77 They are composed of a necrotic caseous center surrounded by a capsule consisting of fibroblasts, epithelioid cells, Langhans giant cells, and lymphocytes. This composition gives tuberculomas a firmness that is different from pyogenic abscesses or malignant gliomas. Calcification can occur in a concentric ring at the margins of the caseous core, as well as at the center of the core, and result in a target sign on CT. Supratentorial tuberculomas are commonly deep, but they can be dural based and mimic meningiomas.78–80 Additionally, cerebellar tuberculomas are frequently in contact with the pia, which makes them difficult to remove without contamination of the subarachnoid space.81
When the caseous core of a tuberculoma liquefies, a tuberculous abscess results. Tuberculous abscesses are usually larger and produce more edema than tuberculomas do. In contrast to tuberculomas, which may not consistently produce positive TB cultures, the liquefied core of an abscess usually contains an odorless, green purulence teeming with acid-fast bacilli. Additionally, abscess walls generally have less granulomatous reaction, although this finding is variable.82 These features give tuberculous abscesses an appearance similar to pyogenic abscesses on imaging studies.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


