Secondary causes of parkinsonism continue to influence our thinking on the possible etiology of Parkinson’s disease (PD) and, as a consequence, assume an importance far greater than their incidence in neurologic practice.
The term secondary parkinsonism refers to bradykinetic syndromes in which a specific cause is known. Separate chapters of this book are devoted to sporadic neurodegenerative parkinsonian disorders, particularly progressive supranuclear palsy (PSP, see Chapter 14), multiple system atrophy (see Chapter 15) and corticobasal degeneration (see Chapter 16).
Several genes have now been linked with monogenetic cause of L-dopa–responsive parkinsonism (1,2). Loss-of-function mutations in four recessive genes (parkin, DJ-1, PINK1, and ATP13A2) cause early-onset parkinsonism, which has a more benign course than PD. Mutations in the two common dominant genes, dardarin LRRK2 and glucocerebrosidase (GBA), increase the risk of developing PD. Dominant missense mutation or locus duplication (or triplication) of the SNCA gene is another rare cause of monogenetic PD (3). Other genetic conditions, particularly Huntington’s disease, spinocerebellar ataxia (SCA) 2, 3 and 17, Wilson’s disease, dopa-responsive dystonia, Perry’s syndrome, familial basal ganglia calcification (Fahr’s disease), C9Orf72 hexanucleotide repeat expansion, fragile X-associated tremor/ataxia syndrome (FXTAS), pantothenate kinase–associated neurodegeneration (PKAN) and other neurodegeneration with iron accumulation (NBIA) conditions, and mitochondrial disorders, including polymerase gamma POLG gene mutation (4), may all present with predominant parkinsonism rather than chorea, ataxia, or dystonia. The identified genes responsible for these heredodegenerative conditions are covered elsewhere in this book.
In this chapter, we have elected arbitrarily to exclude the myriad of rare and esoteric secondary causes in which parkinsonism occurs as part of a more widespread multisystem degeneration and which are only rarely confused with PD. We also have omitted several single-case reports linking infectious diseases, drugs, toxins, and inborn errors of metabolism where cause and effect would benefit from further corroborative reports. Despite these restrictions, the number of secondary causes presenting with PD continues to grow, and space permits only fragmentary accounts of many of the now well-established secondary causes. In addition to the traditional and mandatory divisions relating to cause, we have tried to balance the length of each clinical description to the relative frequency of the causative agent, its contemporaneous interest, the therapeutic opportunity, and the likelihood of achieving accurate diagnosis premortem. For the exotica relegated to tables, the bibliography will provide a stepping stone for additional reading.
IATROGENIC PARKINSONISM
A reversible Parkinson’s syndrome caused by the prescription of antipsychotic dopaminergic antagonist drugs or the dopamine-depleting antihypertensive reserpine has been recognized for half a century, and its early recognition proved to be an important piece in the jigsaw paving the way for the “dopamine miracle” of PD therapy (5). Early descriptions seemed to suggest that neuroleptic-provoked Parkinson’s syndrome was distinguishable from PD by its symmetry and predominantly bradykinetic rigid presentation, as well as a tendency to involve the arms more than the legs (6). Many patients also developed coexisting neuroleptic-provoked dyskinesias, including akathisia and bucco–linguo–masticatory syndromes. However, other studies, particularly in the elderly, have indicated that the clinical picture may be indistinguishable from PD with marked asymmetry and prominent rest tremor (7). A number of these patients have an associated moderate- to high-frequency postural tremor with an additional kinetic component, and a few also present with a strikingly slow 4 to 6 Hz perioral tremor that responds to anticholinergic drugs (“the rabbit syndrome”) (8). In a Department of Elderly Medicine in Scotland study, 51% of 95 elderly new referrals with parkinsonism seen over a 2-year period were found to be receiving neuroleptics (especially prochlorperazine and thioridazine) (9). In a further study in a hospital neurology clinic, 24% of 298 patients were taking drugs implicated in causing parkinsonism (the most common being cinnarizine and sulpiride) (10). Inappropriate prescribing is responsible for a large number of iatrogenic cases, and careful and repeated enquiry about concurrent drug use may be needed to identify an offending medication.
Signs of parkinsonism may start within the first few days of dopaminergic blockade, and the majority of cases are present within 3 months of sustained therapy. Occasionally, severe parkinsonism may occur after sudden concurrent antipsychotic and anticholinergic drug withdrawal, possibly due to cholinergic rebound in the face of persisting dopamine-receptor blockade. In general practice, the antiemetic metoclopramide and prochlorperazine prescribed as a vestibular sedative are common and frequently overlooked causes. There are also reports of generic medications adulterated with neuroleptics causing parkinsonism (11). Anticholinergic drugs are of some symptomatic value when the underlying psychiatric illness precludes neuroleptic discontinuation, but they must be used with great caution in the elderly because of the risk of delirium and amnesia. L-Dopa, on the other hand, is ineffective due to striatal D2-receptor blockade (7). Parkinsonism due to drugs that deplete presynaptic dopamine, such as the antidyskinetic therapy tetrabenazine and reserpine, may be, on the other hand, amenable to treatment with L-dopa (12). If it is possible to stop the offending antipsychotic, recovery usually occurs over several weeks. Occasionally, after depot phenothiazines, recovery may be delayed for many months. There are also reports of spontaneous improvement of neuroleptic-provoked parkinsonism over time despite continuation of the antipsychotic drug, presumably due to the gradual development of striatal dopamine-receptor supersensitivity. However, a few elderly patients fail to recover at all, even after permanent neuroleptic withdrawal when it is usually assumed that PD has been unmasked by pharmacologic dopaminergic blockade. In favor of this is the clinical observation that in some patients iatrogenic parkinsonism may recover, only to be followed a few months later by the emergence of idiopathic PD (13). In patients on long-term neuroleptics who develop parkinsonian symptoms, it is often impossible to distinguish between iatrogenic parkinsonism and emerging PD. Dopamine transporter SPECT imaging is normal in drug-induced parkinsonism, whereas in PD, reduced tracer uptake is demonstrated, indicating presynaptic nigrostriatal dopamine deficit (14). Clozapine and quetiapine are the only widely prescribed antipsychotic drugs that have not been associated with parkinsonism and offer a therapeutic option for schizophrenic patients who develop disabling movement disorders with other antipsychotic drugs. Great care should be taken with antipsychotic therapy, especially in patients over 50. The need for continuing treatment should also be reviewed periodically, and the dose and duration of treatment kept to the minimum needed to effectively control psychiatric symptoms. In elderly patients, the routine coadministration of anticholinergic drugs is inadvisable.
Many patients on long-term antipsychotic drugs manifest subtle parkinsonian signs, but these are usually found on routine examination and do not interfere markedly with psychological rehabilitation. It has been estimated that about 10% to 15% of patients receiving antipsychotics will develop symptomatic parkinsonism. Female gender, old age, high drug potency, and high drug dose are possible risk factors (15), but all these have been contested (16,17). No evidence exists that individuals with a preexisting postural tremor of the hands are at increased risk (18). Patients with neuroleptic-induced parkinsonism do not appear to have a higher incidence of affected first-degree relatives with PD than do age-matched controls (19), but it has been reported that patients with increased echogenicity on transcranial sonography may be at greater risk (20). Patients with dementia with Lewy bodies (DLB) also have been reported to exhibit extreme neuroleptic sensitivity compared with Alzheimer’s disease, leading to the unmasking of cryptic parkinsonism and the potentially fatal neuroleptic malignant syndrome (21).
Although there is a long list of other drugs reported to cause parkinsonism, the mechanisms for most of these are poorly understood, and many are single-case reports. The calcium channel blockers, particularly flunarizine and high-dose cinnarizine, are common causes in some parts of the world (22,23), where they are used as vestibular sedatives and cerebral vasodilators in elderly arteriopaths; dopamine-receptor antagonism is believed to be the likely mechanism of causation. Kava kava, a western Pacific herbal sedative widely used in the West until recent scares of hepatotoxicity can rarely lead to parkinsonism and dystonia (24). Rauvolfia serpentina is one of the 50 fundamental traditional Chinese herbal medicines which has been used for millennia in the Far East as a tranquilizer and antihypertensive. It contains reserpine, an antipsychotic that causes dopaminergic-receptor blockade. We have recently encountered a patient presenting with subacute parkinsonism which completely resolved after discontinuation of the offending remedy. Sodium valproate is a common cause of postural tremor, but a number of convincing cases of reversible Parkinson’s syndrome following chronic administration have now been described (25). Some of these patients have normal dopamine transporter SPECT imaging, suggesting that the cause is unlikely to be presynaptic nigrostriatal dopamine depletion, or the unmasking of PD; some have associated hearing and cognitive deficits (25,26). Although chorea and dystonia are more common side effects with high-dose phenytoin, a few cases of parkinsonism also have been reported (27). A number of anticancer chemotherapy drugs, including cyclophosphamide, busulfan, cytosine arabinoside, vincristine, adriamycin, doxorubicin, and 5-fluorouracil, have been implicated in cases of parkinsonism, but the mechanisms underlying these reports remain obscure (28,29). Immunosuppressants cyclosporine and tacrolimus have also been linked with severe akinetic syndromes (30). Minor concern also continues that benzodiazepines and bupropion, serotonin reuptake inhibitors, central cholinesterase inhibitors, and lithium may aggravate PD (31) (Table 18.1).
VASCULAR PARKINSONISM
Classical “lower-half” Parkinson’s syndrome, associated with confluent subcortical white matter ischemia and lacunar infarcts, is clinically distinct from PD and presents with start hesitation, motor blocking, a broad-based shuffling gait, and falls (47). The speech may be slurred, and some super-added swallowing difficulties, mild arm rigidity, and bradykinesia with slight hypomimia also may occur. Some patients have additional cognitive deficits, pseudobulbar palsy, and pyramidal signs. A clinicopathologic series of 28 cases found that the majority of cases are elderly at presentation and have falls, pyramidal signs, and urinary incontinence (48). Two-thirds had an insidious onset and a relentless rather than stepwise progression of disability. Visual hallucinations were extremely unusual; it was suggested may be a useful differentiating feature from PD (48,49). The diagnosis can be supported by magnetic resonance (MR) imaging, as well as a past history of strokes and vascular risk factors. Dopamine transporter SPECT is usually normal (50,51). A trial of L-dopa up to 1,000 mg daily for at least 6 weeks should always be given, as about a third of patients have a modest improvement. Patients with macroscopic basal ganglia lacunar infarcts, lacunae caused by enlarged perivascular spaces, or neuronal nigral loss are more likely to respond favorably to dopaminergic drugs (52,53) and may have abnormalities on Dopamine transporter SPECT. Bilateral putaminal ischemic or hemorrhagic insults can also cause L-dopa–responsive parkinsonism (54,55).
| Drug-Induced Causes of Parkinsonism |
• Dopamine-receptor blocking drugs (neuroleptics, metoclopramide, prochlorperazine, adulterated generics) (7)
• Dopamine-depleting antichoreic drugs (tetrabenazene) (12)
• Calcium channel blockers (flunarizine, cinarrizine, diltiazem, perhexiline, nifedipine, verapamil) (23,32–36)
• Antiarrhythmic drugs (amiodarone, procaine) (37,38)
• Buspirone, diazepam (39)
• Herbal causes, e.g., kava kava, betel nuts (arecoline), Rauvolfia serpentina (reserpine) (24,40)
• Anticonvulsants sodium valproate, phenytoin (25–27)
• Immunosuppressants (cyclosporine, tacrolimus, busulfan, cytosine arabinoside, vincristine, adriamycin) (28,29,30,41–43)
• α-Methyldopa (45)
• Cephaloridine (46)
• Amiodarone (38)
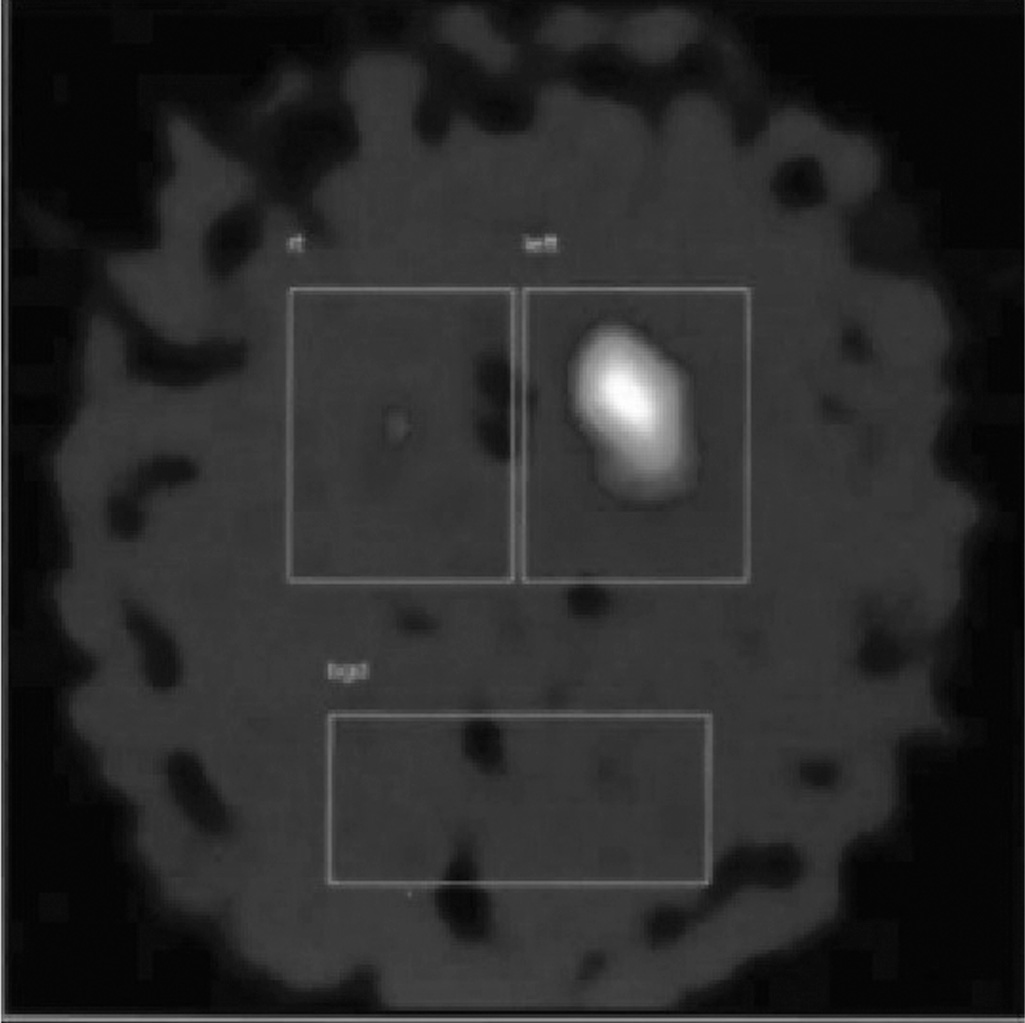
Figure 18.1. Dopamine transporter SPECT showing unilateral loss of dopamine transporter uptake in a patient with vascular parkinsonism following a striatocapsular infarct.
A more uncommon and less well-recognized vascular syndrome may follow an acute striatal infarct with limb hemiparesis (56,57). As the corticospinal tract signs resolve, a bradykinetic rigid syndrome emerges on the same side. Dopamine transporter SPECT may reveal complete absence of uptake on the side of the infarct with complete preservation on the other side (Fig. 18.1). A rest tremor may be present, and the parkinsonism may spread to the other side. A modest response to L-dopa is not uncommon (52,53).
A clinicopathologic investigation was conducted, in which 17 highly selected patients presented with parkinsonism for which no alternative pathologic cause could be found was compared with age-matched controls with comparable vascular risk factors. These cases were identified from an archival pathology collection of more than 600 parkinsonian brains. Microscopic small-vessel disease was more severe in the presumed vascular parkinsonism cases. The majority presented with a “lower-half” Parkinson’s syndrome, but four presented acutely with a hemiparesis and then developed delayed-onset parkinsonism over the following year. These “acute” cases were distinguished pathologically by macroscopically visible lacunar infarcts in regions that could result in reduced thalamocortical drive; intriguingly, four of the patients had additional nigral loss without inclusions. On the basis of this study, operational criteria for the clinical diagnosis of vascular parkinsonism (VP) have been proposed (52,53) (Table 18.2). The use of the University of Pennsylvania Smell Test (UPSIT) also may be a helpful discriminator as olfaction appears to be preserved in VP, whereas 80% of patients with PD are hyposmic (58).
Binswanger disease, a form of leukoencephalopathy caused by hypoxic-ischemia of the watershed periventricular territories, can rarely present as L-dopa–responsive parkinsonism (59). Familial VP and history of migraine should prompt for screening of the Notch3 gene mutations, which cause CADASIL, an autosomal-dominant arteriopathy associated with stroke and dementia (60). Hereditary diffuse leukoencephalopathy with spheroids (HDLS) caused by CSF1R mutations (61) and mitochondrial cytopathies (62) are other differential diagnoses of parkinsonism associated with white matter pathology.
In everyday clinical practice, difficulties surround the relevance in an individual case of extensive subcortical white matter ischemia demonstrated on MR in determining the cause of the parkinsonian syndrome. If the patient responds well and in a sustained fashion to L-dopa (52,53), is hyposmic, and has an abnormal bilateral reduction in dopamine transporter uptake on SPECT, then PD is probable. However, even here, the subcortical ischemic burden may contribute to the phenomenology, such as presence of severe and disproportionate gait, and bulbar and cognitive impairments.
| Possible Criteria for the Clinical Diagnosis of Vascular Parkinsonism |
• Parkinsonism: bradykinesia (slowness of initiation of voluntary movement with progressive reduction in speed and amplitude of repetitive actions in either upper limb or lower limb, including the presence of reduced step length) and at least one of the following: rest tremor, muscular rigidity, or postural instability not caused by primary visual, vestibular, cerebellar, or proprioceptive dysfunction
• Cerebrovascular disease, defined by evidence of relevant cerebrovascular disease by brain imaging (CT or MRI) or the presence of focal signs or symptoms that are consistent with stroke
• A relationship between the two preceding disorders. In practice, (1) an acute or delayed progressive onset with infarcts in or near areas that can increase the basal ganglia motor output (globus pallidus pars externa or substantia nigra pars compacta) or can decrease the thalamocortical drive directly (ventral lateral nucleus of the thalamus, large frontal lobe infarct). The parkinsonism at onset consists of a contralateral bradykinetic rigid syndrome or shuffling gait, within 1 year after a stroke (vascular parkinsonism). (2) An insidious onset of parkinsonism with extensive subcortical white matter lesions, bilateral symptoms at onset, and the presence of early shuffling gait or early cognitive dysfunction
Exclusion criteria for vascular parkinsonism: history of repeated head injury, definite encephalitis, neuroleptic treatment at onset of symptoms, presence of cerebral tumor or communicating hydrocephalus on CT or MRI scan, or other alternative explanation for parkinsonism (52,53)
| Infectious Causes of Parkinsonism |
• Japanese B encephalitis (68,69)
• Encephalitis lethargica (von Economo’s disease) (72,73)
• Coxsackie B virus (74)
• Measles/subacute sclerosing panencephalitis postvaccinal (75–77)
• Poliomyelitis (78)
• St. Louis encephalitis (79)
• Western equine encephalitis (80)
• West Nile virus (81)
• Epstein–Barr virus (82)
• Mycoplasma pneumoniae (41,83,84)
• Syphilis (85)
• Borreliosis (Lyme disease) (86)
• Opportunistic infections causing basal ganglia abscesses (toxoplasma, cryptococcus, mucormycosis) (87)
• Neurocysticercosis (88)
• Tuberculosis (89)
• Malaria (90)
Rare cases of Parkinson’s syndrome also have been reported in association with systemic lupus erythematosus causing cerebral vasculitis or diffuse leukoencephalopathy, which is variably responsive to corticosteroids and immunosuppressive agents (63,64), the antiphospholipid syndrome (65), and moyamoya disease (66).
INFECTIOUS CAUSES
There is copious literature on viral and bacterial causes of parkinsonism stretching back to the celebrated midbrain tuberculomatous “noisette” that invaded the midbrain (67) and led Edouard Brissaud to propose that damage to the substantia nigra might be the principal lesion responsible for parkinsonism. Since then, large numbers of infectious agents have been linked occasionally with an acute transient or reversible parkinsonism (Table 18.3). When the symptoms of parkinsonism develop in the acute or convalescent phase of a febrile illness, an infection is often and reasonably the first etiology to be considered. Blood and cerebrospinal fluid (CSF) should be acquired for culture and virologic study; serum for acute and convalescent titers may be extremely helpful, and polymerase chain reaction (PCR) can be diagnostic if mycoplasma is suspected. Nevertheless, it is common for no transmissible agent to be identified, and the scarcity of reports for some common viral or bacterial pathogens such as measles and mumps render their association with parkinsonism far from convincing (75). The underlying pathologic mechanisms linking infection with parkinsonism are still unclear but it has been proposed that they occur as a direct effect of bacterial or viral infection on dopaminergic function, or as a parainfectious autoimmune process.
Japanese B encephalitis is common in Southeast Asia and affects about 50,000 people a year, and movement disorders are relatively frequent and disabling sequelae. Parkinsonism is usually first noted in the early weeks of the illness as consciousness is regained. Distinctive hypodense abnormalities on T2-weighted MR involving the nigra striatum, pallidum, thalamus, and, sometimes, other brain regions have been described. Prognosis for full recovery must be guarded, and residual disability is relatively common (68,69).
Epidemic encephalitis lethargica (EL or von Economo’s disease) is believed to be caused by an as-yet-unidentified transmissible agent (91). During the 1919-to-1926 pandemic, millions of people died, and, of the survivors, thousands more developed an acute parkinsonism, often indistinguishable from PD. The long-term survivors with postencephalitic parkinsonism (PEP) were found to be exquisitely responsive to low doses of L-dopa but developed severe chorea, respiratory dyskinesias, motor fluctuations, and psychotoxicity in the first few weeks of treatment, causing the drug to be discontinued in most cases (92). Oculogyric crises are a common accompaniment to PEP, and many patients also have associated neurobehavioral disturbances including obsessive–compulsive behavior. The disorder is nonprogressive until old age when some modest deterioration may occur (93). Although the original survivors of the pandemic have almost all died now, sporadic cases of mesencephalitis indistinguishable clinically and pathologically from von Economo’s disease continue to be seen: onset is with drowsiness, delirium, and pyrexia with progression to stupor, sleep inversion, central respiratory disturbances, and coma (72). Studies using PCR to successfully identify the H1N1 influenza virus responsible for the epidemic in 1918 have failed to detect influenza RNA in archival EL and PEP brain tissue (94–96). Enterovirus has been identified in a small number of EL and PEP brains (97). A postinfectious immunologic disorder akin to Sydenham’s chorea has been proposed as an alternative explanation (73,98,99). Studies on patients with sporadic EL have also identified antineuronal antibodies, oligoclonal bands in the CSF, and N-methyl-D-aspartate (NMDA)-receptor antibodies in some cases (72,73,98). MR lesions restricted to the substantia nigra have been demonstrated in acute transient PEP with improvement of bradykinesia correlating with disappearance of the MR lesion (100).
Cases of parkinsonism have been reported in association with West Nile virus, which usually presents neurologically with a meningoencephalitis or an acute flaccid paralysis (81). Mycoplasma pneumoniae is occasionally associated with parkinsonism, which emerges usually as the chest infection is resolving (41,83). An increased risk of PD has also been claimed in poliomyelitis (78).
Human immunodeficiency virus (HIV) can cause Parkinson’s syndrome, either as a result of direct viral damage in HIV encephalopathy (101) or through an opportunistic infection, such as toxoplasma (70) or cryptococcal abscess in the basal ganglia (102). A case of pure parkinsonism as the presenting manifestation of HIV infection in an adult has been reported with mild benefit from L-dopa and complete resolution of motor symptoms following normalization of CD4 count on antiretroviral medications (71). In a Brazilian series of more than 2,000 HIV-positive cases, 28 had movement disorders and 14 of these had parkinsonism. In 12, direct HIV infection involving the nigra was believed to be responsible (103). Of the 169 cases of neurosyphilis in a Chinese University hospital, 7 presented with a movement disorder, and 4 of these have a Parkinson’s syndrome (104). Subacute sclerosing panencephalitis (SSPE) rarely causes parkinsonism, presenting classically with behavioral disturbances, periodic myoclonus, and cognitive deficits. In one case, serial MR images revealed migratory basal ganglia lesions, suggesting axonal spread. The patient improved initially with L-dopa (76). Diagnosis of SSPE hinges on CSF measles antibody titers, oligoclonal CSF banding, reverse transcription PCR, and a distinctive EEG appearance (105).
TOXIC CAUSES
Manganese toxicity played an indirect but important role in the introduction of L-dopa therapy into clinical practice. Cotzias, working at the Brookhaven Laboratories in New York State, studied manganese turnover in Chilean miners who had inhaled ore dust. Most had developed behavioral disturbances after a year or so of exposure, with symptoms of forgetfulness, anxiety, hallucinations, and compulsive behavior, followed months later by parkinsonism and dystonia (106). Manganese was found to accumulate in melanin granules in the substantia nigra, and as a result, Cotzias started to explore ways of augmenting neuromelanin, including the use of high-dose neat racemic dopa (107). Manganese-induced parkinsonism is classically symmetrical in onset with early gait and balance problems, postural tremor, and associated limb dystonia (108). In addition to miners, it has been reported in factory workers making dry batteries (109) and incriminated as the toxin in the fungicide maneb (110). It has also been described in Russian, Estonian, Ukrainian, Latvian, and Georgian drug addicts using ephedrone synthesized from a cocktail, including pseudoephedrine and potassium permanganate (111). Delayed progression of motor symptoms is not uncommon with significant residual deficit and lack of L-dopa response in the majority of ephedrine addicts. High-signal intensities are seen in the striatum, substantia nigra, and globus pallidus on T1-weighted MRI, and together with the normal Dopamine transporter SPECT finding suggest postsynaptic striatal and pallidal damage with sparing of the nigrostriatal dopamine bundle (111,112). Pathologic studies of manganese intoxication have revealed significant globus pallidus and nigra zona reticulata damage (113).
Parkinsonism was reported in 11 out of 51 consecutive patients with severe cirrhosis who were hospitalized to assess their eligibility for hepatic transplantation (114). Presentation was insidious but rapidly progressive, with maximum disability occurring within a year of onset in most cases with early gait disorder and falls and a symmetrical L-dopa–responsive bradykinetic rigid syndrome. Associated focal dystonia was common, but there was no flapping tremor or dementia and Kayser–Fleischer rings were absent. Blood and spinal fluid manganese levels were elevated to the same level as those found in occupational manganese exposure, and the occurrence of parkinsonism seemed more related to the degree of liver failure than its underlying cause; it also appeared independently of any hepatic encephalopathic episodes. Striking hyperintensity was found in the pallidum and nigra on T1-weighted MRI, identical to changes reported in chronic manganese poisoning. Liver transplantation was able to reverse the pallidal MR hyperintensities and restore manganese levels to normal. Patients who die in hepatic coma have a sevenfold increase in pallidal manganese levels, which lends support to the role of elevated brain manganese as the cause of parkinsonism in the chronic hepatocerebral syndrome. Autosomal recessive mutations of the manganese transporter gene, SLC30A10, have recently been linked with a childhood-onset syndrome of parkinsonism, dystonia, and cirrhosis (115). The findings of polycythemia, hypermanganesemia, and radiologic evidence of manganese accumulation in the basal ganglia are useful clinical pointers. Chelation therapy with CaNa2 EDTA can lead to persistent improvement in parkinsonism and dystonia (116). Children on long-term parenteral nutrition, particularly in association with cholestasis, also have been reported to develop hypermanganesemia with parkinsonism and MR basal ganglia signal abnormalities (117). It was claimed that chronic low-level exposure to manganese from welding rods increases the risk of PD (118), but this view has been contested by a meta-analysis which showed the lack of association between welding and manganese exposure and increased risk of PD (119). There also has been concern over the presence of manganese in high-octane fuels (120). The exact mechanisms by which manganese neurotoxicity occurs remain to be determined, but increased oxidative stress in vulnerable brain regions as a consequence of mitochondrial toxicity, and auto-oxidation of dopamine, may be important.
It is now 30 years since the reports of 1-methyl-4-phenyl–1,2,3,6 tetrahydropyridine (MPTP) induced irreversible L-dopa–responsive parkinsonism. The seven cases originally described by Tetrud and Langston were clinically indistinguishable from PD except for their acute-onset and subsequent lack of disease progression (121). All these cases occurred in drug addicts in California who obtained a sloppily prepared supply of the meperidine-related designer narcotic MPPP from a kitchen chemist. It became clear subsequently that MPTP had hit the main distribution line of the northern California drug market and that more than 200 individuals had been exposed. Further epidemiologic tracking over several years led to the identification of a further 22 cases with very soft extrapyramidal signs (122). None of these individuals have presented for subsequent follow-up (Tetrud, personal communication), so the possibility that some may have developed parkinsonism cannot be absolutely excluded. It is interesting to speculate whether the seven index cases received a greater hit of MPTP or whether they were genetically predisposed in some way to the toxin. Three of the patients subsequently underwent fetal mesencephalic implants in Lund, Sweden, but despite marked improvement on F-dopa positron emission tomography (PET) imaging, clinical benefit has in the long term been modest (123). Another patient who was serving a prison sentence underwent deep brain subthalamic nucleus stimulation with a good initial response. Four of the patients became psychotic, and most have continued to abuse drugs, raising the possibility that cross-sensitization to addictive drugs may have been a predisposing factor in the high frequency of psychiatric complications. Freezing, unresponsiveness to L-dopa, and early development of dyskinesias, motor fluctuations, and visual hallucinations were reported in most of the patients. Frontal lobe cognitive deficits have been noted on detailed neuropsychological testing, but dementia has not occurred (Tetrud, personal communication). Three cases have come to autopsy (including two of Langston and Tetrud’s original seven and a later isolated case who also had been independently exposed to MPTP after synthesizing designer drugs). All three had marked nigral neuronal loss, and findings suggestive of an ongoing active pathologic process were noted in two, more than a decade after initial exposure (124). No Lewy bodies or neurofibrillary tangles were identified, and, in contrast to PD, there were no extra-nigral lesions.
A case was reported of a 38-year-old woman in whom reversible parkinsonism was caused by inhalation of heroin pyrolysate (“chasing the dragon”); over 2 months, the woman recovered fully from severe bradykinesia, rigidity, truncal unsteadiness, limited upgaze, and confusion that were associated with severe tetrahydrobiopterin deficiency in the CSF (125).
A report claiming that the serotonergic spree drug ecstasy (MDMA; 3,4-methylenedioxymethamphetamine) could cause severe dopamine neurotoxicity in monkeys prompted intense but short-lived concern, in addition to existing worries about chronic depression. This was supported by the common “street” observation that ravers developed transient twitching and tremors (126) and by one or two isolated reports linking parkinsonism to ecstasy use (127–129). However, fear was allayed when the authors retracted their results, having discovered that all but one of their animals had received erroneously methamphetamine (130). Ironically, shortly before this scare and following a highly publicized British television documentary in which a young stuntman with PD was shown to benefit strikingly from self-medication with ecstasy, global media interest had spurred some interest in ecstasy as a treatment for PD.
A number of industrial toxins also have been linked occasionally with parkinsonism, but the causative mechanisms are largely poorly understood (Table 18.4). Carbon disulphide has been implicated for 150 years as a possible cause of parkinsonism, first in the rubber industry and more recently in silo workers exposed to carbon disulfide fumigant vapors (143). Mercury poisoning is most commonly associated with postural tremor (hatters’ shakes) and psychotoxicity, but parkinsonism can also occur. Of the victims of Minamata disease (methyl mercury intoxication), 8% had parkinsonism, as well as ataxia, neuropathies, and hearing and visual disturbances (144). A few cases of cyanide-induced parkinsonism have been reported (145,147): For example, a 46-year-old geologist who took 1,500 mg of potassium cyanide in a suicide bid became comatose after 15 minutes but was treated promptly with sodium thiosulphate, sodium nitrate, and hyperbaric oxygen. He recovered after 3 days, but over the next 3 weeks, he developed parkinsonism with a shuffling gait, marked bradykinesia, hypomimia, and mild tremor and rigidity, which responded modestly to L-dopa. Neuroimaging showed lesions in the globus pallidus and posterior putamen, and fluorodopa PET study revealed decreased dopa uptake (148). Pesticides, particularly paraquat and rotenone, are associated with increased risk of PD in farmers (149).
| Toxic Causes of Parkinsonism |
• Manganese (miners, welders, methylcyclopentadienyl manganese tricarbonyl in petrol) (106,109,118,131)
• Street drugs of addiction: MPTP, heroin pyrolysate, solvent inhalation (toluene), ecstasy, ephedrone (111,121,125,128,132)
• Aliphatic hydrocarbons (n-hexane and halogens) (122,135,136)
• Glyphosate herbicide (glycine derivate) (142)
• Carbon disulphide fumigant (143)
• Mercury poisoning (144)




