Severe Myoclonic Epilepsy in Infancy: Clinical Analysis and Relation to SCN1A Mutations in a Japanese Cohort
Hirokazu Oguni*
Kitami Hayashi*
Makiko Osawa*
Yutaka Awaya†
Yukio Fukuyama‡
Goryu Fukuma§
Shinichi Hirose§
Akihisa Mitsudome§
Sunao Kaneko||
*Department of Pediatrics, Tokyo Women’s Medical University, Tokyo, Japan
†Department of Pediatrics, Seibo International Catholic Hospital, Tokyo, Japan
‡Child Neurology Institute, Tokyo, Japan
§Department of Pediatrics, School of Medicine, Fukuoka University, Fukuoka, Japan
||Department of Neuropsychiatry, School of Medicine, Hirosaki University, Aomori, Japan
Introduction
After Claes et al (2001) first reported de novo mutations in the Na+ channel gene SCN1A in patients with severe myoclonic epilepsy of infancy (SME), recognition of this rare syndrome, also known as “Dravet syndrome,” has accelerated around the world (1,2,3,4). However, diagnosis of SME is, at times, difficult both clinically and electroencephalographically, in part because of the wide range of clinical and EEG features that change with age and because myoclonic seizures may not be the most predominant seizure type in some patients. The latter observation made one author propose changing its name from SME to “epilepsy with polymorphic seizures” (5). According to the descriptions of Dravet and Dravet et al. (6,7), SME patients have: (1) a high incidence of family history of epilepsy or febrile convulsions; (2) normal development before onset; (3) seizures beginning during the first year of life as generalized or unilateral febrile and afebrile clonic seizures, and (4) the subsequent appearance of myoclonic and often partial seizures. EEGs in the early stage usually do not show paroxysmal discharges, but later generalized spike-waves and polyspike-waves and focal abnormalities appear. Photosensitivity may also appear early. Psychomotor development is initially normal and patients become retarded from the second year of life onward. Ataxia, pyramidal tract signs, and interictal myoclonus usually follow. All seizure types are resistant to all forms of treatment. Thus, the diagnosis of SME largely depends on the combination of clinical and electroencephalographic manifestations that change at different ages. The presence of myoclonic seizures does not appear to be most important despite its terminology.
Because of the lack of strict criteria for inclusion or exclusion, there has been some confusion as to whether patients without myoclonic seizures should be classified as SME, even if other clinical symptoms are identical to that described by Dravet and Dravet et al. (6,7). In Japan, several investigators have become aware of the existence of a distinct group of patients whose clinical findings closely resemble SME. All share most characteristic features of SME, as described by Dravet and Dravet et al. (6,7). However, they do not have myoclonic seizures, an essential diagnostic criterion for typical SME. For this group of patients, other designations have tentatively been offered (8,9,10,11), such as “SME borderline (SMEB),” a “peripheral type of SME,” “infantile refractory grand-mal epilepsy,” or “intractable childhood epilepsies with frequent generalized tonic-clonic seizures.” Finally, the International League Against Epilepsy (ILAE) designated “Dravet syndrome” to include SME and all the SME-related or borderline groups as a part of one syndrome (12). Here, we present various phenotypic manifestations of Dravet syndrome and differences between typical and borderline groups in relation to SCN1A mutations.
Different types of Myoclonic Seizures
The term “myoclonic” in SME leads to some confusion in diagnosis, because it does not specify myoclonic seizures. Instead, it is used in a broader sense to include various myoclonic phenomena (7,12,13). We evaluated patients with SME and identified four types of myoclonic attacks on video-EEG or polygraphic recordings (Fig. 7-1) (13). The first seizure type consisted of symmetrical momentary jerking or twitching of the proximal muscles and eyelids, associated with generalized 3.0–3.5 Hz spike- or polyspike-waves (typical myoclonic seizures according to ILAE classification) (Fig. 7-1A). They tended to repeat and sometimes caused the patient to fall forward or downward. The second type of myoclonic attack consisted of successive myoclonic twitching involving mostly the head and eyelids, resulting in rhythmic retropulsion of the head. In this sense, this seizure type is close to absence seizures with a strong myoclonic component or myoclonic absence seizures (Fig. 7-1B). The first and second types of myoclonic attacks occurred frequently during wakefulness, and were at times provoked by photic or pattern stimulation. The third seizure type was massive myoclonia, which was infrequent and sometimes appeared only before the onset of GTCS, i.e., GCTCS. They were frequently observed during sleep, and considered to be a fragment of GTCS, or an initial component of GTCS (Fig. 7-1C). The final type was nonepileptic segmental myoclonus, often seen during the period of frequent GTCS and later in childhood (Fig. 7-1D). This myoclonus is observed randomly and multifocally in arms and legs and most prominently when the patient is doing purposeful hand movements. Although we did not record the seizures, they appeared as focal or localized myoclonic jerks or attacks as described by other authors (14). They were probably a part of focal motor seizures or part of complex partial seizures or atypical absence seizures (or part of the obtundation status described later) rather than a part of myoclonic seizures in a strict sense.
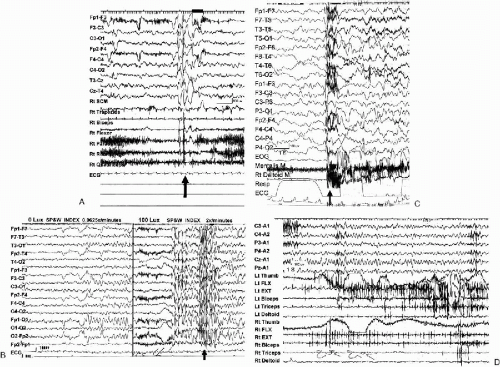 FIG. 7-1. Ictal polygraph of myoclonic seizures. A: Myoclonic seizures. A 9-month-old girl started myoclonic seizures at the age of 5 months. Frequent episodes of myoclonic seizures occurred as single and repetitive jerks. Ictal video shows sudden elevation of both arms, extension of the trunk, and twitching of both eyelids simultaneously twice. B: Successive myoclonic twitching involving mostly head and eyelids. A 2-year 3-month-old girl exhibited marked photosensitivity after the age of 2 years. Attacks were characterized by twitching of eyelids and rhythmic retropulsion of the head for a few seconds. Seizures were easily triggered by photic stimulation, sunlight or a bright room. When she stayed in a dark room (under the lumination of 0 Lux), there were few generalized spike-wave discharges on the EEG (0.0625×/minutes). When she moved to a bright room (constant illumination of 100 Lux), frequent generalized spike-wave discharges were associated with myoclonic seizures (two per minute). Seizures stopped when she moved back to the dark room (constant light sensitivity). C: Massive bilateral myoclonia seizures were observed during all-night sleep recording in a 6-year 5-month-old girl with SME borderland (SMEB). Massive myoclonia or massive spasms (EMG discharges lasted for nearly 1 sec) were associated with diffuse sharp-slow discharge, followed by an arousal response in the EEG. During the all-night sleep recording, three independent massive spasms and one generalized tonic-clonic seizure with a preceding spasm were recorded (13). D: Nonepileptic segmental myoclonus. A 14-year 9-month-old boy with SMEB, developed his first seizure at the age of 3 months. He had had mainly nocturnal GTCS since the age of 6 to 7 years, occurring 6–10 times a month. He also had random and segmental myoclonus that predominantly involved distal muscles. Segmental myoclonus appeared at rest, which increased with purposeful movements. He also showed an ataxic gait. EEG showed 4–5 Hz background slowing without any epileptic abnormality. Myoclonic EMG potentials in both arms often appeared in a tremulous fashion. |
Convulsive Seizures Markedly Precipitated by Elevated Body Temperature
Among the various clinical manifestations of SME patients in Japan, special attention has been paid to the precipitation of seizures by fever and hot baths. Most authors described recurrent episodes of febrile seizures or febrile status epilepticus together with afebrile seizures during infancy and early childhood. These authors emphasized this combination of seizures as one of the characteristic features of the Dravet syndrome (14,15,16,17,18). In our previous study, almost 1/2 of all seizure episodes were associated with elevation of body temperature, when we included seizures during hot bath (19,20). In addition, convulsive seizures often showed polymorphic features at times starting with focal motor seizures with or without secondary generalization, unilateral or alternating unilateral clonic seizures, or other times with generalized clonic or tonic-clonic seizures.
In Japan, hot bath-induced seizures have long received attention as a special type of seizure (21). Since whole-body immersion into a bath tub filled with hot water ranging from 39°C to 42°C for 5 to 10 minutes is a typical custom in Japan, the hot bath-induced seizures were the consequence of the physiological opportunities of raising body temperature in the patients. We recorded the video–EEG during hot-water immersion of children with SME to confirm whether it could provoke attacks or not, and if it did, to establish guidelines for safe bathing (Fig. 7-2A,B) (22,23). It was surprising that clinical overt seizures were provoked in all cases examined after 7 to 25 min of immersion in hot water, when the rectal temperature rose to over 38°. The patients clearly showed an increase in frequency and duration of epileptic discharges which culminated into overt clinical seizures. Interestingly, these seizures included atypical absences, complex partial seizures, and massive myoclonia evolving to GTCS, with elevation of the rectal temperature. Seizures were associated with diffuse irregular spike-wave discharges, culminating into diffuse ictal changes. Myoclonic seizures corresponded to brief diffuse irregular spike-wave complexes. Atypical absence seizures corresponded to longer bursts of diffuse spike-wave complexes. Complex partial seizures (CPS) were associated with more localized and longer bursts of spike-wave discharges. There was no significant difference in the rate of rectal temperature elevation during hot-water immersion between SME patients and three normal controls. Thus, we confirmed that epileptic seizures in SME are very sensitive to body temperature elevation regardless of etiology due to either infection, hot baths, or physical exercise (24).
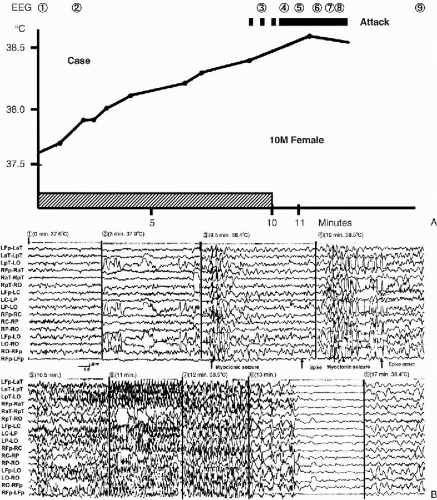 FIG. 7-2. Hot-water immersion: the relationship between rectal temperature and EEG changes in a 10-month-old girl. A: A 10-month-old girl, later confirmed as having SME, had recurrent febrile and afebrile unilateral seizures, partial seizures, and GTCS starting at age 5 months. Rectal temperature was 37.6°C when hot-water immersion started. Rectal temperature gradually elevated to 38.4°C 9 min later, when massive myoclonia developed, as indicated by the dotted horizontal line. Two other massive myoclonic attacks followed 30 and 60 s later, which then evolved to GTCS. B: This EEG was recorded during hot-water immersion. Intermittent diffuse irregular polyspike-wave discharges associated with massive myoclonias developed along with the elevation of body temperature. Massive myoclonias evolved to GTCS when rectal temperature reached 38°C. Seizure threshold seemingly decreased step-by-step as body temperature elevated. There was no difference in the speed of body temperature elevation during hot-water immersion between patients with SME and three age-matched controls. The number corresponded to that in A. Modified from Ref. (23) |
In addition, SME patients are notoriously susceptible to infections and frequently develop hyperthermia during infancy. However, a preliminary immunological study including examination of serum immunoglobulin levels, T-cell function, and complement levels in 16 patients with SME and SMEB failed to identify any immunological abnormality as a cause of susceptibility to infections (25).
Obtundation Status
There are a few special types of seizures observed in SME, which are difficult to classify according to the present International Classification of Seizures. Dravet et al. called them “falsely generalized seizures,” “unstable seizures,” and “obtundation status.” Obtundation status is characterized by prolonged fluctuating disturbances of consciousness with reduced postural tone and some myoclonic jerks (7,12). We recorded the ictal video–EEG in four cases of “obtundation status.” Three of these cases were reported by Fukuyama et al. (26). All cases were characterized by prolonged mild to moderate disturbances of consciousness, at times associated with cyanosis or irregular respiration and/or segmental myoclonus (Fig. 7-3A, B). “Obtundation status” has been associated with various EEG seizure patterns, which are not always identical to the original ones described by Dravet et al. (12). There are at least four EEG patterns associated with obtundation: (1) continuous diffuse dysrhythmia of slow waves, intermixed with focal and diffuse spikes, sharp waves, and spike-waves that are higher in voltage in the anterior and vertex regions (12), (2) continuous posterior localized irregular slow or spike-wave EEG discharges (13,26,28), (3) pseudorhythmic diffuse high-amplitude irregular slow waves, gradually slowing down in frequency (13), and (4) bursts of bilateral, diffuse high-voltage slow waves occasionally notched with small spikes (26,27). This heterogeneity of EEG manifestations is consistent with the polymorphous nature of the seizures in SME. This leads to difficulty in making a diagnosis of SME by diagnosing the major seizure types that characterize the syndrome.
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic Absences: The Seizure and The Syndrome
Myoclonic Absences: The Seizure and The Syndrome
 Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


