Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Charlotte Dravet*
Michelle Bureau*
Hirokazu Oguni†
Yukio Fukuyama‡
Ozlem Cokar§
*Centre Saint-Paul-Hôpital Henri Gastaut, Marseille, France
†Tokyo Women’s Medical University, Department of Pediatrics, Tokyo, Japan
‡Child Neurology Institute, Tokyo, Japan
§Haseki Research and Education Hospital, Department of Neurology, Fatih-Istanbul, Turkey
Introduction
Severe myoclonic epilepsy in infancy (SME) was described by Dravet in 1978 (1). In 1992, there were at least 192 published cases (2). At present, it is more difficult to give a precise figure because the number of publications has greatly increased. In 2002 (3), we counted at least 253 new cases, which represent, totally, about 445 cases. It is interesting to note that most cases are reported from Japan and southern Europe, although SME has been increasingly recognized worldwide, as shown by the description of cases in the United States (4), in China (5), and in Russia (6).
According to the 1989 revised classification of the International League Against Epilepsy (7), this syndrome is characterized by febrile and afebrile generalized and unilateral clonic or tonic clonic seizures that occur in the first year of life in an otherwise normal infant and are later associated with myoclonus, atypical absences, and partial seizures. All seizure types are resistant to antiepileptic drugs. Developmental delay becomes apparent within the second year of life and is followed by definite cognitive impairment and personality disorders. It has been categorized among “epilepsies and syndromes undetermined as to whether they are focal or generalized,” since the syndrome shows both generalized and localized seizure types and EEG paroxysms.
We must underline that many children have been reported by different authors with a similar picture, but without appearance of myoclonias, designated as “borderline SME” (SMEB). Most of them present with association of different seizure types (2,8,9,10,11) whereas others have only generalized tonic clonic seizures (12,13). They can have different EEG features but share the same course and the same outcome as the patients with myoclonias. Thus, they could be included in the same syndrome. This hypothesis seemed to be supported by the genetic studies performed by Doose et al. (13). For this reason, it has been proposed to change its name, first to “epilepsy with polymorphic seizures,” and then to the “Dravet syndrome.” In this chapter, we describe the typical syndrome according to the literature data completed by our present series (105 patients of which 60 selected on the basis of seizures recorded in video-polygraphic EEG have been exhaustively studied) and by the Tokyo Women’s Medical University, Department of Child Neurology (39 patients). The Tokyo study (14) consists of a total of 84 patients, where only 39 patients have the typical form and 45 the borderline form. We shall then discuss the other forms.
General
Epidemiology
SME is a rare disorder with an incidence probably less than 1 per 40,000 (15). Almost the same figure (1/20,000 or 30,000) was reported by Yakoub et al. (10). In these studies, males are more often affected than females, in a ratio of 2:1.
Two authors calculated the percentage of SME in patients with seizure onset in the first year of life: Caraballo et al. (16) found 3% of SME in a series of 471 patients between 1992 and 1997 and Yakoub et al. (10) found 5% among 329 patients between 1980 and 1985. Two others gave a percentage related to the patients with their first seizure before the age of three: 7% (17) and 6.1% (2). In 1999, we find 8.2%. These figures show that this syndrome deserves to be recognized in infants with early convulsions.
Genetics
A large proportion of cases have a family history of epilepsy or febrile seizures (FS). The most usual percentage is about 25%, but it can be higher, up to 53% (18), 57% (15), 64.3% (19), and 71% (20).
Few authors separate FS from epilepsy in the family histories. Dravet et al. (2) mentioned 14% of FS and 18% of epilepsy. Nieto-Barrera et al. (17), in their series of 28 children, indicated 21% of FS and 32% of epilepsy within families. Among them, there were 2 families with both epilepsy and FS.
In the Tokyo study, there were 26% of FS and 13% of epilepsy. In the present Marseille series (60 cases), family antecedents are found in 22 cases (36%): FS in 10 families (16, 6%), epilepsy in 12 families (20%) of which 3 also presented with FS. Parental consanguinity is noted in three cases. The type of epilepsy found in the family is rarely indicated. In some cases, it seems to be an idiopathic generalized epilepsy. In the study by Benloumis et al. (21), patients with SME and those with FS had significantly increased incidence of FS in their relatives compared to those with absence epilepsy and to the control group. The incidence of epilepsy in relatives was higher in patients with SME and in those with absence epilepsy than in the control group, reaching a statistical significance. Epilepsy in relatives of patients with SME had the characteristics of idiopathic generalized epilepsy.
Four pairs of affected monozygotic twins (20,22,23) and one pair of dizygotic twins were reported (24). One other pair has been observed by Santucci M. (personal communication). In the twins published by Musumeci et al. (23), SME was associated with a Rud syndrome, which is an autosomal, recessive disease. Moreover, in two other families, the two siblings (one girl and one boy) were affected (2). In a family studied by Ogino et al. (25), one girl had a typical SME and her sister SMEB.
Recently, several publications reported SME cases in the syndrome of generalized epilepsy with febrile seizures plus (GEFS+) identified by Scheffer and Berkovic (26). Singh et al. (27), among eight probands with SME, found six cases with a family history of seizures. Eighteen members were affected: febrile seizures in eight, febrile seizures + in four, febrile seizures + and partial epilepsy in one, unclassified in two, SME in one, myoclonic astatic epilepsy in one, and Lennox–Gastaut syndrome in one. They conclude that in a significant number of SME cases, a genetic etiology is likely, with most family members having benign epilepsy phenotypes consistent with the GEFS+ spectrum. Veggiotti et al. (28) reported two Italian families of GEFS+ with two siblings affected by SME in each one.
In 2001, Claes et al. (29) found new mutations in the sodium-channel gene SCN1A in all the seven probands with SME that they studied. These mutations were more severe than those observed in the GEFS+ families and occurred de novo. Several other studies have confirmed the finding of SCN1A gene mutations in most of the patients affected by SME, but not in all. The proportion varies from 85.7% (30) to 82.7% (31), 71.4% (32), 44.3% (33), 35% (34), and 33% (35). The differences between inclusion criteria are not sufficient to give a satisfactory explanation of these discrepancies. Only two patients carrying a GABRG2 gene mutation were also reported (33,36). In a large group of 53 subjects negative for SCN1A mutations, the research for GABRG2 mutation was also negative (37). More often mutations are de novo, but they have been detected in one of the parents of 3 patients among 28 tested parents (34), in 2 among 15 tested parents (30), and in the mother of 2 affected siblings (38). Frameshift and nonsense mutations are the most frequent, but missense and others have been reported. They have been shown in the typical as well as in the borderline forms. Correlations between phenotypes and genotypes have been studied without leading to significant differences. At the moment, these results do not allow us to have a clear understanding of the genetic background of the disease, which seems to be heterogeneous. The presence of one gene mutation in a patient supports the diagnosis, but is not yet necessary, as underlined by Scheffer (39).
Personal History
As a rule, SME occurs in normal infants. Significant antecedents were noted in 22% in our series in 1992: intrauterine growth retardation, prematurity, neonatal anoxia, and abnormal pregnancy. This data is in accordance with the literature. Only one author mentioned a percentage of 66%, in 15 patients (40). In one patient, SME was associated with a Rud syndrome (23), in one of our present series with an epidermal naevus syndrome (Solomon syndrome), and in another with a type I neurofibromatosis.
Brain Imaging
Usually, neuroimaging studies do not demonstrate brain lesions, in particular, of the malformative type. However, CT scan and MRI can show signs of slight or moderate, diffuse, cerebral atrophy, cerebellar atrophy, sometimes increased white matter signal (T2 weight) (2,18,40,41). Three times an arachnoid cyst was reported [temporal in the study by Ohki et al. (20), retrocerebellar in our present series]. In some cases, the cerebral atrophy appeared during the course of the epilepsy, while CT scan or MRI were normal at the onset.
Few authors performed interictal SPECT. Nieto-Barrera et al. (18) studied ten patients. SPECT was normal in two cases. It showed areas of hypoperfusion in eight cases, localized in one hemisphere in five, and in both hemispheres in three. Areas of hypoperfusion were concordant with the prevalence of EEG paroxysms in two patients and discordant in two others. Lambarri San Martin et al. (42) studied four patients and found areas of hypoperfusion, temporal in three, and parietal in one.
Ferrie et al. (43) studied eight patients (of which two were considered atypical) by PET scans and MRI. They found normal MRI in all, normal PET in four, but cortical hypometabolism in four. All the latter had lateralized seizures. In one, there was a correlation between focal ictal symptoms and unilateral temporal PET abnormalities. In the three others, the correlation was unclear: asymmetric bitemporal, asymmetric bilateral posterior, marked diffuse hypometabolism.
Semiology at Onset
Seizures began before 1 year in all cases. Initially, prolonged, generalized, or unilateral clonic seizures are typically triggered by fever. Several Japanese authors (8,19,22) underlined the triggering effect of a Japanese style hot water immersion, which produces a body temperature elevation (see below). These febrile seizures tend to be long (more than 20 min) [25% in our present series, 28% in the Tokyo study, 5 cases among 14 for Ohki et al. (20), and 7 among 17 for Yakoub et al. (10)] to recur in clusters in the same day (20) and to evolve into status epilepticus.
However, variations in the onset have been observed by all the authors. Afebrile seizures can occur: 28% (20), 35% in our present series, and 48% in the present Tokyo’s study. In our series these afebrile seizures usually occur in the context of a vaccination or of an infectious episode, or after a bath. Later on, they are associated with febrile seizures in 80% of patients. Nieto-Barrera et al. (18) emphasized the coincidence between the first seizure and the DTP (diphtheria–tetanus–polio) vaccination.
In some patients, isolated episodes of focal myoclonic jerking are noted by parents either some weeks or some days before the appearance of the first convulsive seizure, without any fever and remain isolated, or they occur in the hours preceding the first convulsive seizure, repetitively, together with hyperthermia. Complex partial seizures can also be observed: 2 cases in our present series, at least 5 cases in the Tokyo study, and in 1 case among the 14 reported by Ohki et al. (20). Initial focal seizures are also noted by other authors (44,45).
At this stage of the disease, the EEGs are usually normal. They may display a diffuse or unilateral slowing of the background if they are recorded after a prolonged seizure. In some patients they can show generalized spike-waves elicited by the intermittent photic stimulation (IPS). This early photosensitivity was observed by Dalla Bernardina et al. (41) in 10 cases, by Renier and Renkawek (46) in 1 case, and by Dravet et al. (2) in 13 cases. Rhythmic theta activities at 4–5 Hz can be present in the centroparietal areas and the vertex (40,41).
This first seizure is often considered as a FS, few investigations are performed, and no treatment is given. However, shortly thereafter (2 weeks to 2 months, mean 6 weeks for Dulac and Arthuis, 47), other febrile seizures occur and seizures without fever also appear [2 months to 14 months, mean 5 months for Ohki et al. (20)]. Between 1 and 4 years of age, other seizure types appear, simultaneously with a slowing in the psychomotor development and the picture becomes characteristic of a steady state.
Steady State
Patients with SME have multiple seizure types during the course of the disease: convulsive seizures consisting of either GTCS, generalized clonic seizures (GCS), or alternating unilateral clonic seizures, myoclonic seizures, atypical absences and obtundation status, focal seizures (simple focal motor seizures and complex partial seizures, with or without secondarily generalization), and exceptionally tonic seizures.
Convulsive Seizures
We group under this term all the seizures apparently generalized or unilateral, usually classified as tonic-clonic or clonic. They are present all over the evolution. However, the recent study conducted in Marseille and not yet published, concerning 60 patients whose seizures have been carefully analyzed by video-polygraphic-EEG recordings, demonstrates, in many cases, peculiar clinical and EEG features of these seizures, which do not permit classification among the generalized clonic or tonic-clonic seizures. We have tried to group them by distinguishing several forms: the generalized clonic or tonic clonic seizures (9 patients), the unilateral seizures (9 patients), the “falsely generalized” seizures (10 patients), and the “unstable” seizures (11 patients). In fact, this distinction is somewhat artificial and all these seizures, except the generalized ones, seem to belong to the same category. Furthermore, they can be associated in the same patient. Most of them have been recorded during sleep, either nocturnal or diurnal, at various stages of the evolution, usually after the age of 3.
Generalized Convulsions (Nine Cases)
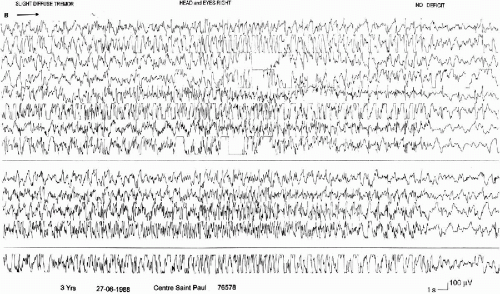
The generalized convulsions (nine cases) evoke the generalized tonic-clonic seizures of the idiopathic generalized epilepsies. However, they are usually shorter, with a very brief tonic phase, with few autonomic symptoms, with a transient postictal flattening quickly replaced by diffuse delta waves. These characteristics are those observed in the GTCS in childhood (Fig. 6-1). In some seizures, the initial tonic phase is almost immediately mixed with the clonic jerks, giving a vibratory aspect which is well documented in the three cases published by Ogihara et al. (48).
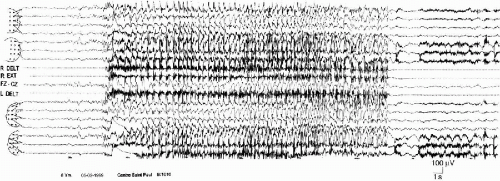 FIG. 6-1. A generalized tonic-clonic seizure in an 8-year-old boy. At onset: brief burst of fast activity (less than 1 s) progressively intermixed with slow waves. The end of the seizure is marked by a brief flattening followed by theta activity. The EMG recorded over the two deltoids shows a tonic contraction followed by sustained myoclonias that become irregular and very fast, with a vibratory aspect. |
True Hemiclonic Seizures
True hemiclonic seizures, corresponding to the description by Gastaut et al. (50), become rare when patients are older. They have been recorded only in 2 young children (16 month and 3 years) in our series of 60 patients. However, there are other unilateral seizures (in seven patients) with different characteristics: shorter duration, association of contralateral tonus changes, ictal EEG anomalies more limited to one hemisphere. They can begin with diffuse spike-waves (49). In all the cases, there are EEG postictal asymmetric signs and, often, a postictal transitory hemiparesia. These seizures can be either on one side or on the other side in the same patient (Fig. 6-2 A, B, and C and Fig. 6-3 A and B). This pattern of alternating unilateral seizures is one of the main characteristics of the syndrome.
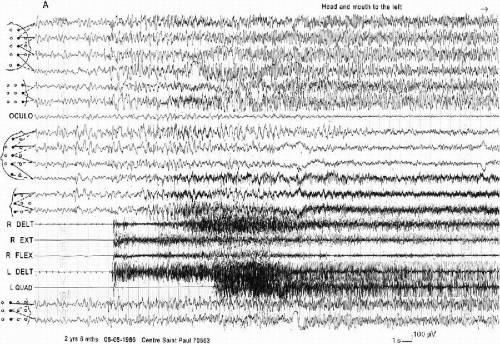 FIG. 6-2. A left hemiclonic seizure recorded in a 2-year 6-month-old boy during a status. A: On the right hemisphere, spikes and slow waves during 5 s, followed by rapid, high-voltage activity; on the left hemisphere, high-voltage spikes and slow waves on the frontocentral area, followed by a low-voltage, rapid activity. Clinically, head and mouth deviation to the left. Polygraphy: diffuse tonic contraction, then vibratory phase on the left muscles. B: Sustained, continuous discharge of slow waves and spikes on the right hemisphere, spreading to the left anterior region, accompanied by rhythmical clonic jerks on the left side of the body. C: At the end, the discharge spreads to the entire left hemisphere. The postictal phase is marked by a depression on the right, associated with a left hemiparesis, immediately followed by a rapid activity, which is the onset of another seizure, whereas delta waves invade the anterior regions of the left hemisphere. |
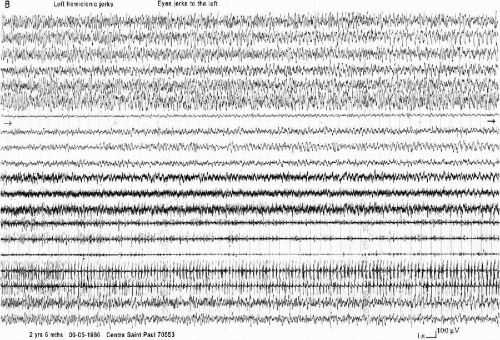 FIG. 6-2. (Contested) |
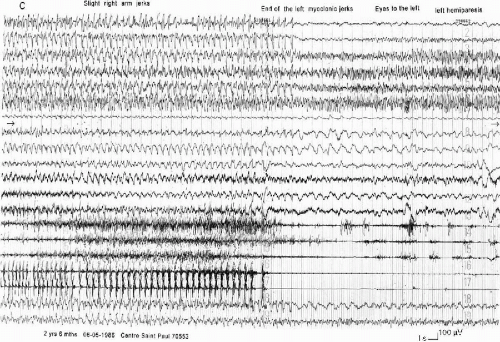 FIG. 6-2. (Contested) |
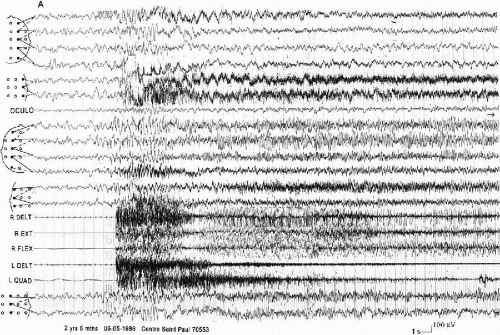 FIG. 6-3. A right hemiclonic seizure recorded in the same boy on the same day. A and B: The aspect is similar to the seizure in Fig. 6-4, but the discharge involves the left hemisphere “as a mirror.” |
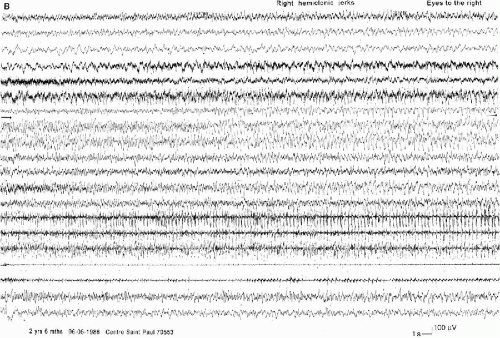 FIG. 6-3. (Contested) |
“Falsely Generalized Seizures” (Ten Cases)
They are characterized by a complicated semiology with some degree of discrepancy between the clinical and the EEG phenomena. The description reported by the parents seems to correspond to a generalized tonic clonic seizure. However, accurate observation through the video-polygraphic EEG recording demonstrates that they are not primarily generalized and they are different from a patient to another one. It is mainly the recording of several muscles on the two sides of the body which permits the analysis of the clinical events. They consist of a bilateral, asymmetric, tonic contraction, leading to variable postures during the seizure (extension of one limb, flexion of another). The onset can be an opening of the eyes preceding the motor phenomena, with or without deviation of the bulbs, the head, and the mouth. The patients seem to be unconscious and do not react to stimuli. Clonic jerks can start in the face or immediately involve the limbs. They are asynchronous, with an asymmetric frequency (vibratory in one side, slower in the other). They often stop on one side and persist on the other, even on a single segment. They last from 30 s to 2 min. The autonomic symptoms are slight: cyanosis, apnea, hypersalivation, and respiratory obstruction occur only at the end of the longest attacks.
The EEG discharge is always bilateral, but according to three modalities. The first consists of bilateral abnormalities from the onset, as a slow spike or a spike-wave, sometimes followed by a brief attenuation, followed by rapid activities and slow waves, still bilateral but more or less asymmetric and asynchronous. In the second, the abnormalities are initially bilateral but become and remain asymmetric during the seizure. In the third, they are bilateral but asymmetric at their very onset. The postictal EEG shows either a diffuse flattening or a diffuse slowing (Fig. 6-4). Sometimes the end of the seizure is not easy to recognize and the child continues to sleep. This asymmetry in the EEGs during the generalized tonic clonic seizures has also been described by others (11,19).
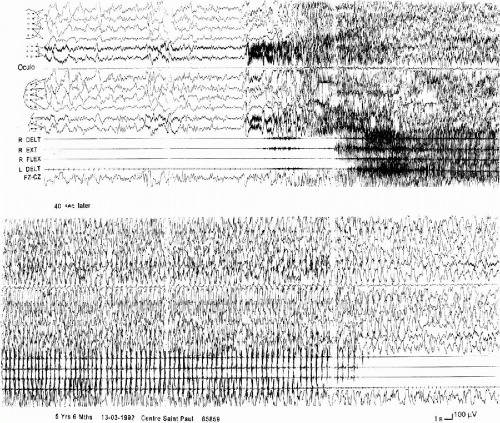 FIG. 6-4. A “falsely generalized” convulsive seizure during sleep in a 5-year 6-month-old boy. Top: At onset a generalized slow spike followed by a low voltage theta activity, then bilateral fast activity of high voltage. Note the muscular artefacts on the EEG, which correspond to the facial onset of the clinical seizure. The EMG recording of the upper limbs shows a slight initial tonic contraction which begins 2 s after the EEG onset, remits, and then is followed by an irregular tonic contraction associated with a vibratory aspect. Bottom: 40 s later, diffuse spike-waves associated with bilateral myoclonias, which become progressively asynchronous and stop on the left deltoid before they stop on the right muscles. At the end of the seizure, note the absence of a flattening, but the presence of high-voltage delta waves. |
“Unstable Seizures” (11 Cases)
These seizures are characterized by the topographic changes of the ictal EEG discharge in the same seizure. The clinical manifestations are near that of the “falsely generalized” seizures with asymmetric and asynchronous tonic and clonic movements, sometimes predominant on one side or shifting from one side to the other. However, the EEG discharge involves irregularly different parts of the brain. It can start in one localized area of one hemisphere, then spread either to the entire hemisphere, either asymmetrically to the two hemispheres, or to another area of the same hemisphere or of the opposite hemisphere, then return to the first involved hemisphere. The end of the discharge can occur either in this hemisphere or contralaterally (Fig. 6-5 A and B). The ways of propagation are very variable from one seizure to another in the same patient and even in the same recording. The relationship between the clinical events and the accompanying EEG is not always clear.
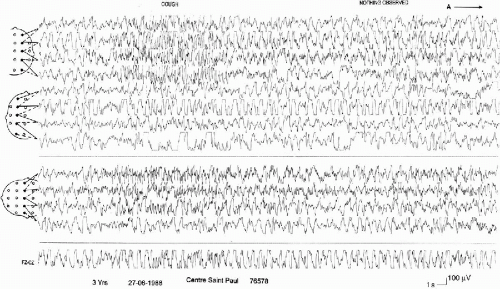 FIG. 6-5. An “unstable convulsive seizure” occurring during slow sleep in a 3-year-old boy. A: Brief diffuse lowering of voltage intermixed, on the right hemisphere, with rapid activity, then spikes and slow waves evident only on the right hemisphere, during 10 s, followed by a more or less rhythmic activity around 10 Hz on the right centroparietal area. B: Continuation of the seizure: 20 s after the onset of that activity, a similar one appears on the left frontocentrotemporal region, progressively associated with slow waves on the left hemisphere, becoming slower at the end of the seizure. Clinically, only a slight diffuse tremor and a head and eyes deviation to the right are observed. |