Chapter 25 Sleep and Host Defense
The Acute Phase Response and Host Defense
Enhanced sleep in response to infections occurs in the context of dozens of other responses, collectively termed the APR. Recent advances in innate immunity research provide a framework for understanding many of the shared mechanisms underlying the APR in general and sleep regulation specifically. The APR is a critical innate immune response1 that follows any inflammatory challenge, such as an infection or traumatic injury. Local inflammatory challenges, for example, minor cuts or bruises, may activate a low-level APR that is not perceived by the victim. But when the challenge is severe enough, it activates a full-blown systemic APR within 24 hours and the patient feels sick. In the case of infections, the function of the APR is to wall off and destroy invading microorganisms, to remove tissue debris, and to alert the host to the invasion so that systemic protective responses are mobilized. The systemic inflammatory response activates the brain, liver, and bone marrow to react in a stereotypic manner. The APR has both behavioral (fever, excess sleep, anorexia, etc.) and biochemical markers (C-reactive protein, serum amyloid A, mannose binding protein, etc.). Increased secretion of a broad array of endocrine hormones, including the stress hormones, also occurs. This complex of responses leads to host protective behaviors (such as social withdrawal that avoids predation),2 physiological responses (such as fever, which can thermally inhibit growth of some microorganisms),3 and immune responses (such as mobilization of leukocytes and natural killer (NK) cells.)1 Hormonal changes (such as prolactin regulation of antimicrobial nitric oxide levels)4 and biochemical changes (such as potentiation of microbial phagocytosis)5 also contribute to host defense. The APR is the first line of defense against infections and the trigger for acquired immunity mediated by specific antibodies and cytotoxic T lymphocytes needed to clear the infection.6
A major class of protein mediators, the cytokines, initiates the APR.7 Cytokines are generally associated with immune cells, but they are made by all cells. Over 100 of these intercellular signaling molecules have been identified, and the complexity of their interactions rivals that of the nervous system. Cytokines induce themselves and other cytokines, and they form biochemical cascades characterized by much redundancy. Cytokines are classified into two major groups, type I cytokines that promote inflammation (proinflammatory) and type II cytokines that suppress it (antiinflammatory).8 Three proinflammatory cytokines, specifically interleukin (IL)-1β, tumor necrosis factor-α (TNF-α), and IL-6, appear to be primary triggers of the APR, including the situation when excess NREM sleep occurs.7,9 The class II cytokines such as interferon (IFN)-α, IFN-β, IL-4, and IL-10 downregulate the APR and may also modulate the sleep response; for example, IL-4 and IL-10 inhibit spontaneous NREM sleep (Fig. 25-1). Cytokines can act in an autocrine, juxtacrine, paracrine, or endocrine manner to activate numerous APRs via such effectors as nitric oxide, adenosine, and prostaglandins.7,9,10
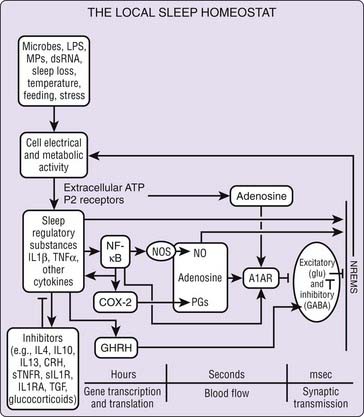
Figure 25-1 Local sleep homeostat. IL-1β and TNF-α are part of the brain cytokine network that includes several other endogenous somnogenic substances and sleep inhibitory substances. Cell electrical and metabolic activity, induced by multiple stimuli including inflammatory signals, is associated with ATP release. ATP via purine P2 receptors causes the release of IL-1β and TNF-α from glia. They, in turn, activate NFκB. Redundant pathways as well as negative feedback signals involving additional cytokine and hormones provide stability to the sleep regulatory system as well as alternative mechanisms by which a variety of sleep promoting, or sleep inhibitory, stimuli may affect sleep. Our current knowledge of the biochemical events involved in sleep regulation is much more complicated than that shown here. For example, multiple additional cytokines (e.g., acidic fibroblast growth factor) enhance sleep.10 TNFR, soluble TNF receptor; anti-TNF, anti-TNF antibody; NGF, nerve growth factor; NFκB, nuclear factor κB; IL1RA, IL1 receptor antagonist; sILIR, soluble IL1 receptor; anti-IL1, anti-IL1 antibody; CRH, corticotropin releasing hormone; PG, prostaglandin; COX-2, cyclooxygenase-2; TGF, transforming growth factor; NOS, nitric oxide synthase; A1R, adenosine A1 receptor; GHRH, growth hormone releasing hormone; (→) indicates stimulation; (—) indicates inhibition.
A major breakthrough in our understanding of the APR is the recognition that all known microorganisms have one or more biologically stable and chemically unique structural components, or they produce one during replication.11 These components are termed pathogen-associated molecular patterns, or PAMPs. These PAMPs are recognized by a specialized group of membrane-bound or intracellular receptors (pathogen recognition receptors, or PRRs)6 that include the Toll-like receptors (TLRs) and the nucleotide-binding domain and leucine-rich repeat domain receptors (NLRs) (more commonly designated as nucleotide-binding oligomerization domain, or Nod, proteins.)12 The PRR binding of microbial PAMPs induces cytokines, and these cytokines in turn upregulate PRRs and cytokines in neighboring tissues, resulting in amplification of the response. Thus in infectious illness, microorganisms induce cytokines, then cytokines activate the APR and thereby facilitate host defense via dozens of protective mediators and activated immune cells.1,7 Enhanced non–rapid eye movement (NREM) sleep appears to be one outcome of this cytokine cascade in infectious illness.
Sleep Changes After Infectious Challenge
Viral Challenge
Viral diseases that cause central nervous system lesions and/or systemic inflammation can affect sleep.13 In von Economo’s seminal paper14 he related the location of brain lesions in encephalitis lethargica to changes in sleep patterns.* This work led to the concept that sleep was an active process, not simply the withdrawal of sensory stimuli, and to the idea that there was some degree of localization of neural networks regulating sleep. Despite the importance of this work, many years passed before the direct effects of viral infections on sleep were experimentally determined. During the early stages of human immunodeficiency virus (HIV) infections, before AIDS onset, patients have excess stage 4 NREM sleep during the latter half of the night.16 Another central nervous system viral disease, rabies, is also associated with disrupted sleep.17 In such diseases, it is difficult to distinguish whether the effects of the viral infection on sleep are direct or whether they result from virus-induced brain lesions.13 Influenza virus localizes to the respiratory tract during the early stage of disease and has been used in several sleep investigations. Smith and colleagues18 at the British Common Cold Unit reported that low doses of influenza in humans induce excess behavioral sleep and cognitive dysfunction; these symptoms appear after low viral doses that fail to induce the better known characteristics of the APR, such as a fever. However, polysomnography was not used in this study. More recently, animal studies of influenza viral infections clearly show that sleep is profoundly affected by infectious challenge. The sleep changes in influenza-infected mice,19 illustrated in Figure 25-2, are similar to those seen in bacterial infections (described hereafter). In a rabbit model, intravenous injections of influenza virus are also associated with large increases in NREM sleep, although the virus fails to replicate in this species.13
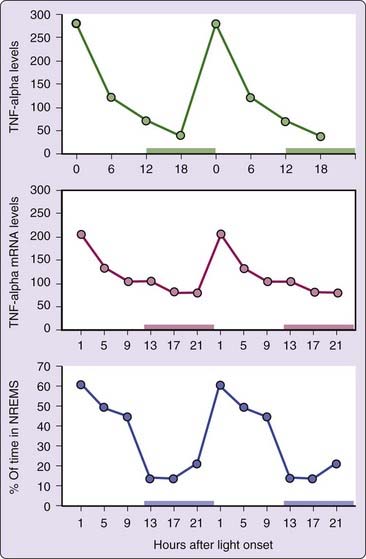
Figure 25-2 Influenza virus challenge induces prolonged increases in NREM sleep (top panel) and decreases in REM sleep (bottom panel) in mice. Data are from reference 19. Filled triangles represent data collected after viral inoculation. Open triangles represent the averaged baseline data collected over a 3-day period just prior to viral inoculation. Horizontal dark bars show lights-off periods for each experimental day.
The influenza-infected mouse is more applicable to humans because mouse-adapted strains of this virus can be inoculated via the respiratory tract and can fully replicate in the lungs, causing a severe APR. Mice challenged intranasally with influenza virus display profound increases in NREM sleep lasting 3 or more days, while rapid eye movement (REM) sleep is inhibited7,13 (see Fig. 25-2). The macrophage appears to the critical immune cell type driving increased NREM sleep, whereas NK cells, neutrophils, and T lymphocytes do not play a significant role.20 Different strains of mice respond differentially to viral challenge21 indicating a strong genetic component affecting the sleep response to influenza virus. Genetic regulation of the inflammatory response to influenza in mice and humans has been reviewed.22
One generic viral PAMP that induces excess sleep and other APRs is virus-associated double-stranded (ds) RNA. All viruses examined to date produce virus-associated dsRNA, which is generally derived from the annealing of viral replication products rather than from the virus itself.23 Virus-associated dsRNA is recognized by the PRR TLR3, as well as intracellular helicases, and dsRNA induces numerous cytokines such as IL-1, IL-6, TNFα, and IFN.23 Virus-associated dsRNA can be extracted from lungs of infected mice24 and is capable of inducing an APR in rabbits similar to that of live virus. Similarly, rabbits given short double-stranded (but not single-stranded) oligomers that correspond to a portion of influenza gene segment 3 also exhibit large increases in NREM sleep.7 Synthetic dsRNA (polyriboinosinic : polyribocytidylic acid, or poly I : C) induces an APR virtually identical to that of influenza virus when inoculated into the lungs of mice primed with IFN-α. Poly I : C administered to rabbits also induces an influenza-like APR, but the corresponding single strands of poly I and poly C are inert. In rabbits, poly I : C can substitute for virus in the induction of hyporesponsive state to viral challenge. These observations7,23 suggest that virus-associated dsRNA is sufficient to initiate the APRs seen in influenza-infected mice. Mice deficient in TLR3 demonstrate attenuation of influenza APRs.23a
Rabbits challenged with viable virus or poly I : C have increased plasma antiviral activity that occurs concomitantly with the changes in sleep. The antiviral activity is attributed to IFN-α and other cytokines. Injection of IFN-α into rabbits also induces sleep responses similar to those induced by virus, poly I : C, or the double-stranded viral oligomers.25 High doses of IFN-α also stimulate NREM sleep in other species,26 but lower doses that simulate levels of IFN-α seen during an infection inhibit both NREM and REM sleep in humans.27
Interferons have long been thought to play a major role in viral (and poly I : C) symptoms.23 A tool now widely used by researchers to better understand the role of a specific cytokine or hormone in host defense is the so-called knockout (KO) mouse.7 These mice have a single gene deletion, and comparing their responses with wild-type mice provides valuable insights into contributions of a single gene product, such as type I IFN, to the APR. Mice genetically deficient for the receptor that binds both IFN-α and IFN-β (the type I receptor) still respond to poly I : C challenge with excess sleep and a hypothermic response similar to that seen in infected wild-type mice. However, the APR occurs earlier in influenza-infected IFN-receptor KO mice,28 suggesting that type I IFNs may modulate the APR by regulating proinflammatory cytokine production. Influenza-infected IFN-receptor KO mice are less ill in the later phase of the infection and appear to recover sooner.28 Our current hypothesis is that cytokines (other than IFNs) that are known to be somnogenic are likely to mediate the excess sleep response to influenza virus. For example, influenza virus challenge of mice deficient in both the 55-kD and 75-kD TNF receptors reduces EEG delta power, a measure of sleep intensity, whereas in wild-type controls delta power increases.29 Duration of NREM sleep and REM sleep changed to the same extent in both strains after viral challenge. Another mediator that plays a role in both sleep and host defense4 is nitric oxide synthesized by multiple nitric oxide synthetases (NOSs).30 Mice deficient in either neuronal NOS or inducible NOS have attenuated NREM sleep responses to influenza challenge compared to infected wild-type controls.30
Mice and rats with natural mutations of the growth hormone releasing hormone (GHRH) receptor express a dwarf phenotype and altered spontaneous NREM sleep.31 The GHRH receptor is a candidate gene for regulation of NREM sleep increases in response to influenza virus,13 and its expression is upregulated in the hypothalamus by IL-1β. If dwarf mice with nonfunctional GHRH receptors (called lit/lit mice) are infected with influenza virus, then they fail to show the increase in NREM sleep or EEG delta power seen in heterozygous controls.32 Instead, infected lit/lit mice manifest a pathological state with EEG slow waves, enhanced muscle tone and increased mortality.32 Such results indicate that single genes can substantially modify sleep responses to infectious challenge and suggest that the sleep responses forming part of the APR at least correlate with survival.
Influenza virus has been a popular model for acute phase studies because it has been assumed that the virus does not invade the brain or lead to the complications associated with neurovirulent viruses. Recent studies in our laboratory, however, demonstrate that the strain of influenza most commonly employed in acute phase studies rapidly invades the olfactory bulb of the mouse brain following intranasal inoculation.33 The virus activates microglia in the outer layer of the bulb and induces significant IL-1β and TNF-α upregulation at the same time (15 hours post-infection) that the APR (hypothermia, in this case) begins.33 These studies suggest that cytokines made in the olfactory bulb could impact the APR to influenza virus.
Bacterial Challenge
Sleep responses are also observed after bacterial challenge. Indeed, results obtained after challenging rabbits with the gram-positive bacteria Staphylococcus aureus were the first to suggest that NREM sleep responses formed part of the APR.34 In those experiments rabbits were given S. aureus intravenously to induce septicemia; within a few hours of the inoculation large excesses in NREM sleep were observed. Associated with the increase in NREM sleep were increases in amplitude of EEG slow waves. EEG slow wave (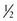 to 4 Hz) amplitudes are thought to be indicative of the intensity of NREM sleep. This initial phase of increased duration and intensity of NREM sleep lasted about 20 hours; it was followed by a more prolonged phase of decreased duration of NREM sleep and decreased EEG slow wave amplitudes.34 During both phases of the NREM sleep changes, REM sleep was inhibited and animals were febrile. Other changes characteristic of the APR, for example, fibrinogenemia and neutrophilia, occurred concurrently with the changes in sleep.34
to 4 Hz) amplitudes are thought to be indicative of the intensity of NREM sleep. This initial phase of increased duration and intensity of NREM sleep lasted about 20 hours; it was followed by a more prolonged phase of decreased duration of NREM sleep and decreased EEG slow wave amplitudes.34 During both phases of the NREM sleep changes, REM sleep was inhibited and animals were febrile. Other changes characteristic of the APR, for example, fibrinogenemia and neutrophilia, occurred concurrently with the changes in sleep.34
In subsequent studies in which gram-negative bacteria and other routes of administration were used, a similar general pattern of biphasic NREM sleep responses and REM sleep inhibition was observed.7,13 However, the timing of sleep responses depends upon both the bacterial species and the route of administration. For example, after intravenous administration of Escherichia coli, NREM sleep responses are rapid in onset, but the excess NREM sleep phase lasts only 4 to 6 hours. The subsequent phase of reduced NREM sleep and reduced amplitude of EEG slow waves is sustained for relatively long periods. In contrast, if the gram-negative bacterium Pasteurella multocida is given intranasally, a different time course of sleep responses is observed. In this case, the increased NREM sleep responses occur after a longer latency and the magnitude of the increases in NREM sleep is less than the effects of this pathogen given by other routes of administration. P. multocida is a natural respiratory pathogen in rabbits.
The intestinal lumen of mammals contains large amounts of bacteria, some of which leak into the intestinal lymphatics under normal conditions. Changes in intestinal permeability by inflammatory challenges increase the release of bacterial products into the lymphatics. Local lymph node macrophages phagocytose and digest these bacterial products,9,35 releasing somnogenic PAMPs as described later. This mechanism is viewed as operating at a low basal rate under normal conditions and is amplified greatly during systemic inflammation. It is also likely to be involved in sleep responses induced by sleep deprivation and excess food intake (discussed hereafter). Reduction of bacterial populations in the intestine is associated with a reduction of sleep.36
A specific muramyl peptide derived from bacterial cell wall peptidoglycans was isolated from the brain and urine of sleep-deprived subjects.37 This muramyl peptide was the first bacterial PAMP to be associated with sleep regulation.37 Peptidoglycan components are recognized by certain NLRs7 and appear to play a major role in the pathogenesis of inflammatory mucosal diseases.7 The sleep promoting activity of muramyl peptides is dependent upon their chemical structure.38 Many muramyl peptides are also immune adjuvants and pyrogenic, although the structural requirements for these biological activities are distinct from those required for sleep-promoting activity.7,38
Another bacterial cell wall product that is involved in sleep responses that are induced by gram-negative bacteria is the lipopolysaccharide component of cell wall endotoxin. Lipopolysaccharide is the dominant PAMP associated with endotoxin, it binds to TLR4,7 and has been intensively studied with respect to sleep effects in both animal models10 and humans.39 Lipopolysaccharide and its toxic moiety, lipid A, are somnogenic in animals and man.7,38 Modification of the lipid A structure alters somnogenic activity (e.g., conversion of diphosphoryl lipid A to monophosphoryl lipid A) and reduces its somnogenicity. Humans inoculated with lipopolysaccharide and followed for 12 hours manifest sleep changes, fever, cytokine expression, and hormonal changes39 somewhat similar to those seen in animals, though EEG changes are distinct from those seen in rabbits or rats and NREM sleep increases require a higher dose of lipopolysaccharide than does REM sleep reduction.
Other microbes, for example, fungal organisms such as Candida albicans or protozoans such as Trypanosoma brucei brucei express their own PAMPs, bind to specific TLRs, and also have the capacity to induce sleep responses.40 Some of these microbe-induced sleep responses are quite interesting. Trypanosomiasis in rabbits is associated with recurrent bouts of enhanced NREM sleep occurring about every 7 days. Trypanosomes undergo antigenic variations in the host; the proliferating new antigenic variants stimulate the host immune response, and such periods are accompanied by excess NREM sleep.40 Like bacteria and viruses, fungi and protozoans have the capacity to enhance cytokine production by the host.
In summary, infectious challenge is associated with profound changes in sleep. As mentioned in the overview of the APR, PRRs such as the TLR and NLR receptor families detect the various somnogenic PAMPs described previously and provide an overarching theory that explains why diverse microbial factors activate stereotypic host defense responses such as fever, anorexia, and excess sleep.7 These microbe-induced sleep responses are now considered part of the APR, and, like the other components of this response, sleep may be adaptive.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


