Chapter 117 Sleep-Related Cardiac Risk
Abstract
In healthy individuals, sleep is usually salutary and restorative. Ironically, during sleep in patients with respiratory or heart disease, the brain can precipitate breathing disorders, myocardial ischemia, arrhythmias, and even death. Our observation that 20% of myocardial infarctions (MIs) and 15% of sudden deaths occur during the period from midnight to 6:00 AM projects to an estimated 300,000 nocturnal MIs and 48,750 nocturnal sudden deaths annually in the U.S. population.1 The latter figure is equivalent to 87% of the number of U.S. fatalities due to automobile accidents and is more than 2.5 times the number of U.S. deaths resulting from human immunodeficiency virus infection. Thus, sleep is not a fully protected state. Furthermore, the nonuniform distribution of deaths and MIs during the night is consonant with provocation by pathophysiologic triggers. The two main factors implicated in nocturnal cardiac events are sleep state–dependent surges in autonomic activity2 and depression of respiratory control mechanisms,3 which affect a vulnerable cardiac substrate. Precise characterization of their interaction in precipitating nocturnal cardiac events is, however, incomplete. Although sudden death during sleep can be presumed to be painless, in many cases it is premature because it occurs in infants and adolescents and in adults with ischemic heart disease, for whom the median age is 59 years. Populations at risk for nocturnal cardiorespiratory events include several large patient groups (Table 117-1). For example, atrial fibrillation is common in sleep apnea.1a–1f
Table 117-1 Patient Groups at Potentially Increased Risk for Nocturnal Cardiac Events
| INDICATION (U.S. PATIENTS/YR) | POSSIBLE MECHANISM |
|---|---|
| Angina, myocardial infarction (MI), arrhythmias, ischemia, or cardiac arrest at night; 20% of myocardial infarctions (~300,000 cases/yr) and 15% of sudden deaths (~48,750 cases/yr) occur between midnight and 6:00 AM. | The nocturnal pattern suggests a sleep state–dependent autonomic trigger or respiratory distress. |
| Unstable angina, Prinzmetal angina | Nondemand ischemia and angina peak between midnight and 6:00 AM |
| Acute MI (1.5 million) | Disturbances in sleep, respiration, and autonomic balance may be factors in nocturnal arrhythmogenesis. Nocturnal onset of MI is more frequent in older and sicker patients and carries a higher risk of congestive heart failure. |
| Heart failure (5.3 million) | Sleep-related breathing disorders are pronounced in the setting of heart failure and may contribute to its progression and to mortality risk. |
| Spousal or family report of highly irregular breathing, excessive snoring, or apnea in patients with coronary disease (5 to 10 million patients with apnea). | Patients with hypertension or atrial or ventricular arrhythmias should be screened for the presence of sleep apnea. |
| Long QT syndrome | The profound cycle-length changes associated with sleep may trigger pause-dependent torsades de pointes in these patients. |
| Near-miss or siblings of victims of sudden infant death syndrome (SIDS) | SIDS commonly occurs during sleep with characteristic cardiorespiratory symptoms. |
| Brugada syndrome in Western populations; Asians with warning signs of sudden unexplained nocturnal death syndrome (SUNDS) | SUNDS is a sleep-related phenomenon in which night terrors may play a role. It is genetically related to the Brugada syndrome. |
| Atrial fibrillation (2.5 million) | Twenty-nine percent of episodes occur between midnight and 6:00 AM. Respiratory and autonomic mechanisms are suspected. |
| Patients on cardiac medications (13.5 million patients with cardiovascular disease) | Beta-blockers and calcium channel blockers that cross the blood–brain barrier may increase nighttime risk because poor sleep and violent dreams may be triggered. Medications that increase the QT interval may conduce to pause-dependent torsades de pointes during the profound cycle-length changes of sleep. |
It is an insidious component of the problem of nocturnal risk that many people are unaware of their respiratory or cardiac distress at night and therefore take no corrective action. Thus, sleep presents unique autonomic, hemodynamic, and respiratory challenges to the diseased myocardium that cannot be monitored by daytime diagnostic tests. The importance of nocturnal monitoring of patients with cardiac disease extends beyond identifying sleep state–dependent triggers of cardiac events because nighttime myocardial ischemia, arrhythmias, autonomic activity, and respiratory disturbances carry predictive value for daytime events (Box 117-1).
Box 117-1 Predictive Value of Nocturnal Cardiorespiratory Status
 Because parasympathetic nerve activity is elevated during sleep in healthy individuals, lack of circadian pattern of heart rate variability and baroreflex sensitivity may be readily monitored for increased risk of cardiac events.
Because parasympathetic nerve activity is elevated during sleep in healthy individuals, lack of circadian pattern of heart rate variability and baroreflex sensitivity may be readily monitored for increased risk of cardiac events.




In this chapter we discuss the pathophysiologic mechanisms responsible for sleep-related cardiac morbidity and mortality. For a review of mechanisms and treatment of nocturnal arrhythmias, see Chapter 118. Effects of sleep-disordered breathing and apnea on the cardiovascular system are discussed in Chapter 119.
Autonomic Activity and Circulatory Function During Sleep
The generalized decrease in mean heart rate and arterial blood pressure at the onset of sleep and throughout non–rapid eye movement (NREM) sleep, which occupies 80% of sleep time, has prompted the assumption that sleep is a period of relative autonomic inactivity. NREM sleep, the initial stage, is characterized by marked stability of autonomic regulation with a high degree of parasympathetic neural tone and prominent respiratory sinus arrhythmia.2,4 Baroreceptor gain is high and contributes to the stability of arterial blood pressure and to overall cardiovascular homeostasis.5 Muscle sympathetic nerve activity is stable, falls with the transition from awake to NREM sleep, and decreases progressively with depth of sleep,2,6 reaching half the awake value during stage 4.2 Short-lasting increases in muscle sympathetic nerve activity, heart rate, and arterial blood pressure accompany the appearance of high-amplitude K-complexes during stage 2.2,6 Heart rate accelerations may even precede the electroencephalographic arousals of stage 2 and REM sleep.7 During transitions from NREM to REM sleep, bursts of vagus nerve activity may result in pauses in heart rhythm and frank asystole. Transitions between REM and NREM sleep elicit posture shifts that are associated with varying degrees of autonomic activation and attendant changes in heart rate and arterial blood pressure.8 These shifts in body position increase in frequency as individuals age and sleep becomes fragmented.
Autonomic nervous system activity is dramatically altered when REM sleep is initiated (Fig. 117-1). REM sleep is marked by profound muscle sympathetic nerve activation, in terms of both frequency and amplitude,2,6 which attains levels significantly higher than in wakefulness.2 Sympathetic nerve activity is concentrated in short, irregular periods that are most striking when accompanied by intense eye movements.2 These bursts trigger intermittent increases in heart rate and arterial blood pressure to levels similar to those in wakefulness, with increased variability.2,6,7 Significant surges and pauses in heart rate during REM sleep have been described in several species, including humans.6,7 Cardiac efferent vagal tone and baroreceptor regulation5 are generally suppressed during REM sleep, and breathing patterns may become highly irregular and may lead, in susceptible individuals, to oxygen desaturation. Thus, while subserving the neurochemical functions of the brain, REM sleep can disrupt cardiorespiratory homeostasis. The brain’s increased excitability during REM sleep can also trigger major surges in sympathetic nerve activity to the skeletal muscular beds, accompanied by muscular twitching,2 which interrupts the generalized skeletal atonia of REM.8 The peripheral autonomic status characterized by muscle sympathetic nerve recording is compatible with reduced neuronal activity in the brainstem and other regions of the brain and reduced cerebral blood flow during NREM sleep and, during REM sleep, with increased brain activity in several discrete regions to levels higher than waking values.9
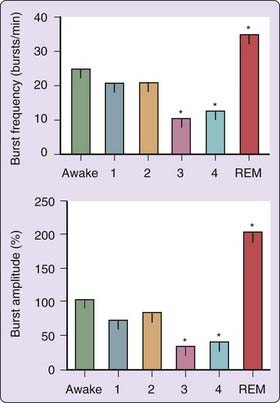
The decline in autonomic activity during sleep is also evident in peroneal muscle sympathetic nerve activity2,6 and peripheral levels of epinephrine and norepinephrine, and mirrors the generalized sleep-induced decline in heart rate and arterial blood pressure.10 A nocturnal nadir in plasma catecholamines is evident at 1 hour after sleep onset. Plasma cortisol is also depressed during sleep; increased levels are initiated at 5:00 AM.
In the absence of readily achieved, direct measures of cardiac-bound nerve activity, analysis of heart rate variability (HRV) has emerged as a widely accepted method for measuring cardiac sympathetic versus parasympathetic neural dominance. High-frequency (HF) HRV is a general indicator of cardiac parasympathetic tone and includes the effects of respiration. The low- to high-frequency (LF/HF) ratio is widely accepted as an approximation of cardiac-bound sympathetic nerve activity, as validated by studies involving beta-adrenergic receptor blocking agents. Decreased HRV, indicating a decline in parasympathetic nerve activity, is an established indicator of risk for sudden cardiac death after MI. HRV analysis reveals a generalized increase in vagus nerve activity and a decrease in cardiac sympathetic nerve activity across the sleep period,11,12 probably reflecting the dominance of total sleep time by NREM sleep. HRV studies using 5-minute intervals provide results consistent with muscle nerve recording, indicating increased HF and decreased LF (or parasympathetic nerve dominance) in NREM sleep, but decreased HF and increased LF (or predominant sympathetic nerve activity) in REM sleep and during wakefulness.7 In healthy individuals, the increase in HRV measures of cardiac sympathetic nerve activity at onset of REM sleep is initiated before7,12 the transition from NREM sleep as classically defined from the polysomnographic record.
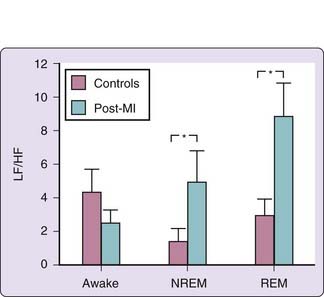
The typical circadian pattern of decreased nocturnal cardiac sympathetic nerve activity as described by heart rate and HRV studies is altered in patients with coronary artery disease,13,14 MI,11,15 and diabetes,16 suggesting either increased nocturnal cardiac sympathetic nerve activity or decreased parasympathetic nerve activity compared with healthy subjects. The HF component has been observed to decrease approximately 10 minutes before onset of nocturnal myocardial ischemia.14 In unmedicated patients with a recent MI, the LF/HF ratio was significantly increased during both REM and NREM sleep, in contrast to healthy subjects, in whom this ratio during REM sleep is similar to awake levels and higher than during NREM sleep (Fig. 117-2).11 The conclusions were reached that MI decreases the capacity of the vagus nerve to be activated during sleep, resulting in unbridled cardiac sympathetic nerve activity,11 and that loss of rise in the HF component is characteristic of patients after an MI and with residual myocardial ischemia.15
Nocturnal Cardiovascular Events
Nocturnal Myocardial Ischemia and Angina
Accurate assessment and treatment of nocturnal angina has been a subject of concern for more than 2 centuries. Heberden in 1768 described angina that “will often oblige [the patients] to rise up out of their bed every night for many months altogether.” John Hunter, the well-known 18th-century surgeon, reported chest pains that “seized him in his sleep so as to awaken him.”17 As early as 1923, MacWilliam18 postulated that the mechanisms of nocturnal ventricular fibrillation and angina were stimulation of sympathetic nerves and increased arterial blood pressure. He described “reflex excitations, dreams, nightmares, etc., sometimes accompanied by extensive rises of arterial blood pressure (hitherto not recognized), increased heart action, changes in respiration, and various reflex effects” and noted “the suddenness of development of the functional disturbances in arterial blood pressure, heart action, etc., in the dreaming state.” He documented greater stress on the circulatory system during dreaming than during wakefulness, with arterial blood pressures reaching 200 mm Hg. In the middle of the 20th century, the renowned cardiologists Paul Dudley White and Samuel Levine remarked on the frequency of MI and angina in sleep and suggested an association with dreams.
Ischemic activity is an important prognostic marker in patients with cardiac disease, and characteristics of both REM and NREM sleep may conduce to nocturnal myocardial ischemia and angina. The few studies in patients with cardiac disease that have used sleep staging have concluded that in the absence of significant depression of left ventricular function, nocturnal ischemic events occur primarily during REM sleep,19,20 which is characterized by increased sympathetic nerve activity, metabolic demands, and heart rate surges. In patients with stable coronary artery disease, myocardial ischemia is largely attributable to bouts of sympathetically mediated surges in heart rate and resultant metabolic demands in flow-limited, stenotic coronary arteries.4,14,20–24 Nowlin and coworkers20 attributed nocturnal angina to heightened blood pressure after performing detailed, multisession polysomnographic analysis of four patients with advanced coronary artery disease and nocturnal angina pectoris. They established that attacks of nocturnal angina occurred predominantly during REM sleep (32 of 39 recordings) and were associated with heart rate acceleration. Dream content, in patients who could describe it, included awareness of chest pain and involved strenuous physical activity or emotions of fear, anger, or frustration.
Nocturnal myocardial ischemia may be generated by mechanisms in addition to sympathetic nerve activity and unsatisfied metabolic demands. This possibility is suggested by the finding that nighttime ischemic events remain, although they are less frequent, in patients receiving beta-adrenergic receptor blockade therapy, the primary therapy that effectively reduces the overall incidence of and suppresses the morning peak in cardiac events by containing sympathetic nerve activity and demand-related myocardial ischemia.24,25 The main factors that may contribute to nondemand-related myocardial ischemia during NREM sleep are decreased coronary perfusion pressure as the result of hypotension4,24–27 and increased coronary vasomotor tone.26 These influences decrease the metabolic threshold for induction of nocturnal myocardial ischemia, which has a nadir between 1:00 AM and 3:00 AM.21,26,28 During these hours in patients with stable coronary disease, Benhorin and colleagues26 observed that myocardial ischemia can be provoked at heart rates of 83 beats per minute (bpm), in contrast to 96 bpm during midday, and that its incidence was not affected by beta-adrenergic receptor blockade. Patel and colleagues25 noted that nocturnal myocardial ischemia is attended by heart rate elevations of 6 bpm or less in patients with unstable angina receiving beta-adrenergic receptor blocking agents. Mancia4 hypothesized that the hypotension of NREM sleep is a major contributor to nocturnal myocardial ischemia and MI because it “reduces the volume and velocity of blood flow, favoring the development of thrombi and embolic and ischemic phenomena before and after arousal.” It has also been postulated that myocardial ischemia provoked by transient thrombus formation29 is attributable to the nocturnal nadir in endogenous fibrinolytic activity,29 as well as to peaks in serum levels of plasminogen activator inhibitor29 and tissue plasminogen activator antigen, increasing blood viscosity or hypercoagulability at night, and free-radical generation.
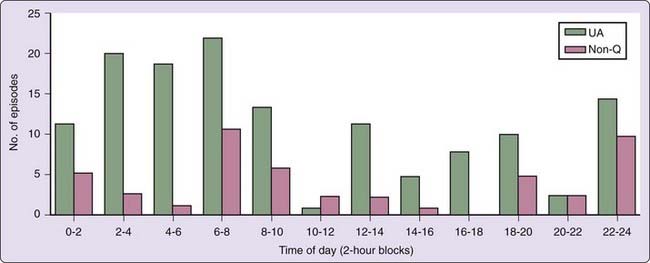
Nondemand nocturnal myocardial ischemia is prevalent among patients with more severe coronary disease,24,28,30 acute coronary syndromes,15 or diabetes, populations with significant endothelial dysfunction. Indeed, it has been concluded that nondemand nocturnal ischemic episodes disclose a critical underlying coronary lesion, coronary vasospasm, or transient coronary artery stenosis.25 Patel and colleagues25 documented a nocturnal peak in ischemic events in their study of 256 hospitalized patients with the acute coronary syndromes of unstable angina and non–Q-wave MI (Fig. 117-3). Electrocardiograms were recorded within hours after patients’ admission for chest pain to the coronary care unit for new-onset angina, sudden acceleration of previously stable angina, or angina within 1 month of MI. In hospital, they received optimal medical therapy aimed at containing demand-related myocardial ischemia. It is important to note, however, that the peak in out-of-hospital onset of the syndromes followed the usual circadian pattern, as reported by Cannon and coworkers31 in the Thrombosis in Myocardial Infarction (TIMI) III Study of 3318 patients. By contrast, in patients with longstanding diabetes or with documented autonomic nervous system dysfunction, there is no nocturnal decrease in myocardial ischemia or onset of acute MI.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


