Chapter 27 Spinal Cord Injury
• Spinal cord injury (SCI) is a problem in both the developed and developing countries and is associated with significant morbidity and long-term human and financial costs.
• Spinal cord injury is a medical emergency. Patients suffering from this injury should be treated in specialized centers with strict adherence to proper clinical examination, imaging, and medical and surgical therapies.
• SCI is a result of primary damage to neural structures and vasculature, followed by secondary damage from physiological insults such as hypotension and hypoxia and resultant cytotoxic swelling and death of neurons and glial cells.
• There have been a multitude of strategies of neuroprotection and repair in SCI and can be found in Box 27.2.
• Spinal shock is represented by depressed or absent spinal reflexes below the injury site. Neurogenic shock after trauma results from disruption of the sympathetic nervous system usually at T6 or higher, which may lead to hypotension and may be difficult to distinguish from hypovolemic shock. The optimal treatment of neurogenic shock is restoring intravascular volume; if symptoms of neurogenic shock persist, vasopressors such as dopamine may be useful.
• Central cord syndrome is often the result of a traumatic spinal cord contusion and affects the central part of the spinal cord. This can cause loss of bilateral pain and temperature sensation and loss of motor function at the level of the lesion and below.
• Early closed reduction of bilateral locked facets in an awake patient, who can participate in a neurological examination, may relieve spinal cord compression and restore normal alignment.
• The use of methylprednisolone as a neuroprotective agent in acute SCI shows moderate benefit in terms of neurological outcome but the benefits of its administration should be considered against the risks associated with medical comorbid conditions in individual patients.
• There are several indications for surgical decompression in SCI. The first is to treat spinal instability caused by fractures and ligamentous injury that may cause further neurological damage. There is some evidence, albeit controversial or evolving, that surgical decompression within 24 hours may improve neurological outcome in some patients with isolated SCI, especially those with cervical SCI who are deteriorating neurologically.
To frame the concepts of SCI—from pathophysiology to translational research to clinical practice—it is important to understand the reasons for undertaking such an endeavor. Spinal cord injury has been studied at a population level in order to understand the incidence, prevalence, and economic costs of this devastating event. These studies tend to come from developed countries that have the financial resources to conduct such research, but this problem certainly does not respect borders. In fact, the frequency of neurotrauma in the developing world is estimated to be higher than in the developed world for reasons such as poor quality of roads, unsafe driving practices, old vehicles without seatbelts or airbags, and a greater proportion of relatively inexpensive transportation such as small motorcycles. Nonetheless, the estimated incidence in the developed world is in the range of 11.5 to 53.4 people per 1 million population.1 This number must be interpreted with caution as some authors point out that the actual incidence may be on the higher side because an unknown number of patients suffering SCI die at the scene of the accident without ever being examined or treated in a hospital.2 Another study estimated that approximately 48% to 79% of individuals die either at the scene of the accident or on arrival to hospital.3 This study was performed in the mid-1970s and most surgeons treating this disease would agree that the number of patients who die is likely less today, mainly as a result of an improved understanding of the cardiorespiratory complications that can arise following injury and advanced intensive care units that are well versed in managing these complications.
One way to comprehend the costs involved in treating a person with SCI is to imagine that a 25-year-old sustains an injury and becomes completely dependent on family and the health care system from this point forward in life. One study estimated that the total cost would be in the range of $3 million (U.S. dollars),4 not to mention the impact of this event on the person’s family and peers. The average age of SCI patients is the late 20s and early 30s and tends to occur more commonly in males. Going back to our example, this event therefore affects people as they are starting their careers and families. A lack of income combined with insurmountable medical costs translates into financial and emotional hardship. Depression and forms of psychological distress are common in this patient group. In well-developed health care systems support networks exist to aid patients and families through the life that they could not have imagined. In third world countries, patients and families are left to their own devices. From either case comes a drive to understand the pathobiology of this devastating condition and hopefully offer treatment strategies. In the pages that follow, we will systematically discuss the events that occur following a traumatic accident—from the shear forces that destroy white matter tracts to the cellular mechanisms that result. With this as a foundation we will discuss different translational research programs that are under way to minimize neurological deficit and hopefully offer neuroregenerative strategies in the near future. Lastly we discuss how a detailed understanding of the pathophysiology of this condition has led to clinical treatment strategies. These strategies are an essential knowledge base for any medical student or resident interested in the treatment of this devastating condition.
Pathophysiology
Acute traumatic injury to the spinal cord can come in the form of a blunt mechanism, such as an acrobat falling onto a hard surface or a person thrown from a car during an automobile collision, or a penetrating injury such as a stab or gunshot wound. The forces involved in each of these events are transmitted to the spinal column and if great enough result in disruption of the bony and ligamentous structures and in damage to the neural elements. Damage to the spinal cord or the exiting nerve roots can result in motor, sensory, or autonomic dysfunction. In an attempt to delineate the precise cause of neurological dysfunction researchers have divided the temporal sequence of destructive events into primary and secondary injury. Primary injury refers to the destructive forces that directly damage the neural structures such as the shear force tearing an axon or direct compressive force occluding a blood vessel, resulting in ischemia. These destructive primary mechanisms not only result in instantaneous damage to neurons and blood vessels but also initiate a cascade of cellular mechanisms that result in ongoing damage to the neural structures, termed secondary injury. In fact, in cases of ongoing primary injury, for example, in the setting of a fracture dislocation where the bony spinal column is displaced and physically pushed up against the spinal cord, these cellular mechanisms are thought to be locked into the “on” position until such physical forces are removed either by closed reduction or surgically. Secondary injury may persist from hours to weeks to years following primary injury. A great deal of work has gone into the detailed understanding of these cellular cascades and along with this work has come an appreciation for the destructive effects of the mechanisms and the role of potential therapeutics to halt these cascades. In the remainder of this section we will highlight the most important secondary mechanisms of injury (Fig. 27.1) and in the section titled “Translational Research” we will discuss some of the therapeutic targets that resulted from these basic science discoveries.
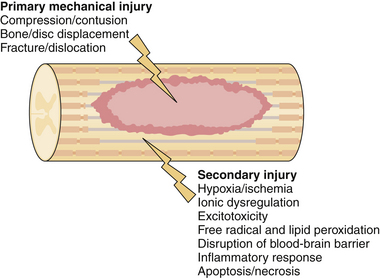
Secondary injury stems from any aberrant physiological or physical force and results in further tissue damage and potential exacerbation of primary injury. Two of the easiest physiological parameters to recognize are hypotension and hypoxia. Both of these conditions are relatively common in trauma patients, who often sustain concomitant injuries and blood loss. Both are also amenable to medical management shortly after the patient arrives at the hospital. Specific treatment options will be discussed later; here we highlight the importance of these two causes of secondary injury and the fact that they further exacerbate secondary cellular cascades. Both global hypotension and hypoxia add to the overall effect of hypoperfusion to the damaged area of the spinal cord. Several other mechanisms add to hypoperfusion including disruption of the microvasculature, loss of normal autoregulatory mechanisms, and increased interstitial pressure.5–7 The effects of this ischemic insult are cytotoxic cell swelling of both neurons and glial cells; as a downstream effect of this, blockade of action potentials has been demonstrated as a result of axonal swelling.8
Ionic dysregulation and excitotoxicity are closely linked events that contribute to tissue damage. Calcium dysregulation in particular has been widely linked to cell death through a number of different processes including calpain activation, mitochondrial dysfunction, and free radical production.9 Whether calcium dysregulation or disruption of other ionic gradients occurs, loss of ionic homeostasis is central to both necrotic and apoptotic cellular death. Excitotoxicity results from activation of glutamate receptors via the influx of both sodium and calcium ions. As cell death occurs, either through the process of necrosis or apoptosis, extracellular glutamate levels rise presumably due to the failure of energy-dependent transporters such as the Na+/K+-ATPase membrane transporter.10 In the next section we will discuss how antagonists of NMDA (N-methyl-D-aspartic acid) receptors are potential targets of translational research in order to minimize cellular death.
Free radical–mediated secondary injury occurs through the process of lipid peroxidation that ultimately results in disruption of axons and death of both neurons and glial cells. Considering the mechanism of free radical damage, peroxynitrite and other reactive oxygen species act through a chain reaction that ultimately destroys cellular membranes and leads to cell lysis, organelle dysfunction. and calcium dysregulation.11 This, in turn, adds to the ionic dysregulation described in the preceding paragraph. Free radicals have been measured in the damaged tissue of experimental models and are found to be elevated for approximately 1 week following injury, returning to preinjury baseline only after 4 to 5 weeks.12 Examples of these free radicals include hydrogen peroxide, hydroxyl radical, nitric oxide, and the superoxide radical. Specific radicals, such as peroxynitrite, have been directly liked to the activation of apoptotic cascades in rat SCI models.13 As with NMDA receptors, free radical oxygen species become a potential target of translational research. If one is theoretically able to mediate the level of these dangerous compounds, could one not prevent the degree of secondary damage that occurs following SCI? This idea will be expanded on in the translational research section below.
Tight junctions between endothelial cells act as an interface between the systemic circulation and the central nervous system. Known as the blood-brain barrier (BBB), it also functions in the spinal cord and acts as a selective barrier that allows passage of solutes such as glucose while maintaining impermeability to macromolecules and infectious agents such as bacteria. Endothelial cells and astrocytes are normally bridged by tight junctions and are interspersed with very selective transmembrane proteins to allow the passage of vital molecules and ions. With the destructive forces that accompany SCI comes the destruction of the BBB. This can result either from direct mechanical destruction, such as damaged blood vessels and spinal cord parenchyma, or from destruction at a cellular level. The latter is best understood as a result of the effects of numerous inflammatory mediators on the endothelial cells that disrupt the normally impermeable barrier. One way researchers have studied this effect is through the use of a compound called horseradish peroxidase. When injected into the peripheral circulation of a normal animal, this compound does not cross the fortress of the BBB and remains in the systemic tissues. When injected into animal models of SCI, the compound was found to permeate into the CNS at a peak time of 24 hours following injury. Such permeability was persistent for up to 2 weeks following injury.14 A number of compounds have been identified that are thought to contribute to this ongoing permeability following injury, each of which is related to the secondary mechanisms of SCI. Inflammatory mediators affecting vascular permeability include tumor necrosis factor-α (TNF-α) and interleukin 1β (IL-1β) and both have been shown to be unregulated following SCI.15,16 Other compounds that play a role include matrix metalloproteinases, histamine, and reactive oxygen species including nitric oxide.12
The inflammatory reaction following SCI has received a renewed interest in recent years largely because of what is thought of as the dual nature of the immune response. On the one hand inflammatory mediators seem to be responsible for the ongoing destruction of tissue, and on the other hand they seem to be clearing cellular debris and optimizing the environment for regenerative growth. Several noncellular mediators are involved in the cascade, the most prominent of which are TNF-α, interferons, and interleukins.17 Cellular mediators following SCI include both resident microglia and peripheral inflammatory cells. A few of the well-characterized examples, mainly from studies in the rodent population, include astrocytes, T cells, neutrophils, and invading monocytes.18 A great example of this dual nature of protection and destruction comes from studies involving TNF-α, which has been demonstrated to be significantly unregulated following SCI.19 Subsequent studies involved the application of neutralizing antibodies to TNF-α following experimental injury, resulting in improved neurological function.20 However, TNF-α-deficient mice have been shown to have higher numbers of apoptotic cells, increased lesion size, and worse function following SCI when compared to wild-type mice.21 This demonstrates the complexity of the inflammatory cascade in SCI and should point the novice reader in the direction of thinking beyond the notion of one type of molecule causing destruction; rather, one must view the many players as acting in a complex pattern, some of which may aid in optimizing function and others that may result in ongoing destruction. The role of research in this field is to tease out these details and provide specific targets for translational research that aim to optimize the potential for recovery.
Cellular death via the apoptotic pathway is thought to play little role in the fate of neurons22 but a considerable role in the fate of oligodendrocytes23 following SCI. Although it may be difficult to demonstrate the apoptotic mechanism of neuronal death in human SCI, there is some evidence for its role in animal models.24 In contrast, the apoptotic mechanism of oligodendrocyte death has been convincingly demonstrated and its mechanism at least partially elucidated. Following SCI, microglia are activated within the first 2 to 48 hours and express the Fas ligand.25 This ligand has a receptor largely expressed on the oligodendrocyte, and communication occurs via the p75 neurotrophin receptor.26 The interaction of the Fas ligand and its receptor initiate apoptosis by way of the caspase cascade that ultimately results in proteolysis, DNA cleavage, and cellular death. This pathway has been the target of translational research strategies and will be expanded on in the next section.
In contrast, cellular death via the necrotic pathway has been demonstrated in both the neuronal and non-neuronal cell populations. Several of the secondary mechanisms of injury previously discussed, such as hypoperfusion and excitotoxicity, culminate in the necrotic death of neurons. Oligodendrocytes are also susceptible to this death pathway and the loss of this cell type has the consequence of axonal demyelination. This demyelination, in combination with other insults on the neuronal processes such as lipid peroxidation and ischemic swelling, results in death of the associated neuronal cell bodies.27 Animal studies have provided evidence that preserved axons represent a critical therapeutic target in order to regain neurological function.28 In stark contrast to this, postmortem human studies have failed to identify demyelinated axons29 and may represent an important disconnect between animal and human pathology following SCI.
Translational Research
As previously stated, an in-depth understanding of secondary mechanisms of SCI has led to identification of numerous possible targets for prevention of this secondary damage and possible regeneration of partially damaged neural circuits. Here we will review a number of agents that are currently under investigation. Broad categories include neuroprotective agents (minocycline and riluzole), myelin-associated inhibitors of neural regeneration (ATI355 and Cethrin), and cellular transplantation strategies (activated autologous macrophages, bone marrow stromal cells, and human embryonic stem cells). This list is by no means exhaustive. Numerous other strategies have been attempted in the past and many lessons have been learned—both positive and negative. Readers interested in a recent history of translational research in the field of SCI are referred to a recently published open source review.30 Alongside developments in neurobiology have come advances in clinical management and operative strategies. In a similar fashion to the scrutiny of drug delivery trials, these clinical and surgical options have also been the focus of clinical trials and extensive review to determine if their effects provide better outcomes for patients. Research that has gone into blood pressure management, methylprednisolone therapy, closed reduction of bilateral locked facets, and early surgical decompression will be discussed in the following section on clinical management.
Neuroprotective Agents
Several promising neuroprotective strategies are under investigation (Box 27.1); two of these are reviewed in greater detail here. Minocycline is a tetracycline derivative that has been of interest to neuroscience for quite some time. It has been shown to have neuroprotective properties in a diversity of animal models, including those that aim to study stroke, Parkinson’s disease, Huntington disease, amyotrophic lateral sclerosis (ALS), and multiple sclerosis.31 As a result of its promise in these conditions, its potential application was carried over to the realm of SCI. Initially studied in animal models, its benefit became apparent in improved neurological outcomes32 and its mechanism of action was thought to be a result of decreased microglia activation and antiapoptotic pathways mediated by the inhibition of cytochrome c release. Because of its extensive use in other neurological conditions, and promise in animal models of SCI, at least two clinical trials are under way to test the efficacy of minocycline in human SCI.
< div class='tao-gold-member'>
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


