aSpecial circumstances of the early and incipient stages for which early anesthetic therapy should be considered are listed in Table 38.2.
The premonitory stage consists of confusion, myoclonus, or increasing seizure frequency, the early stage consists of a continuous seizure, and the refractory stage may consist of subtle generalized convulsive SE or NCSE. It has now become clear that the “transition” stage from the early to the late stage of SE is not fixed in time and may vary depending on the underlying etiology. SE should not be considered refractory if therapy has been inadequate.
A predictable sequence of EEG progression occurs during the clinical stages in experimental models and humans: (i) discrete seizures with interictal slowing, (ii) waxing and waning of ictal discharges, (iii) continuous ictal discharges, (iv) continuous ictal discharges punctuated by flat periods, and (v) periodic epileptiform discharges (PEDs) on a flat background (Fig. 38.1) (23,24). However, every episode of SE does not pass through every one of these defined stages (25) (Fig. 38.2). The PED stage may also consist of either lateralized or bilateral patterns (23). The response to treatment appears to depend on the electrographic stage (see below: Trends in Patients with Status Epilepticus). In one study (23), discrete seizures were all controlled with diazepam (six of six patients); whereas in the PED stage, the seizure stopped in only one of six patients and overt clinical seizures were converted to either subtle or electrographic seizures in the five of six patients.
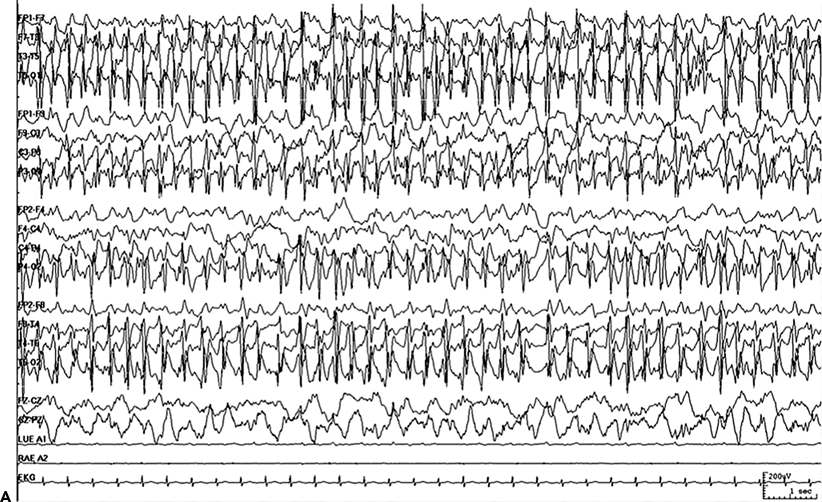
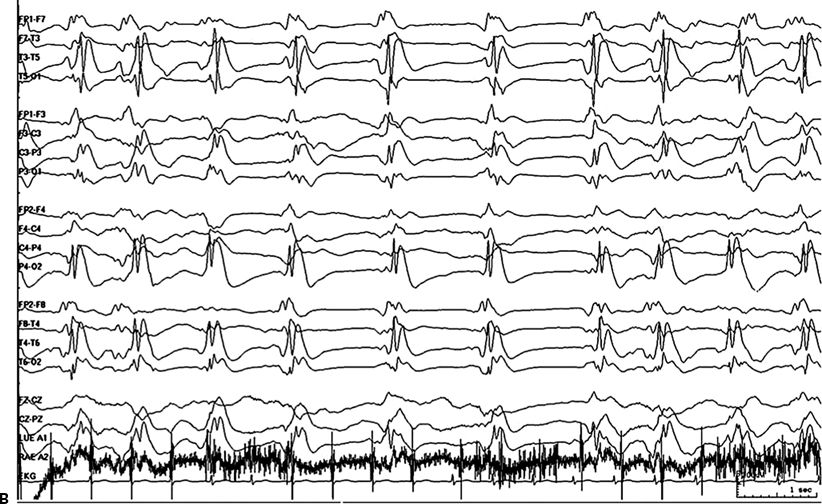
Figure 38.1. A:Continuous ictal discharges. B:PEDs on a flat background.
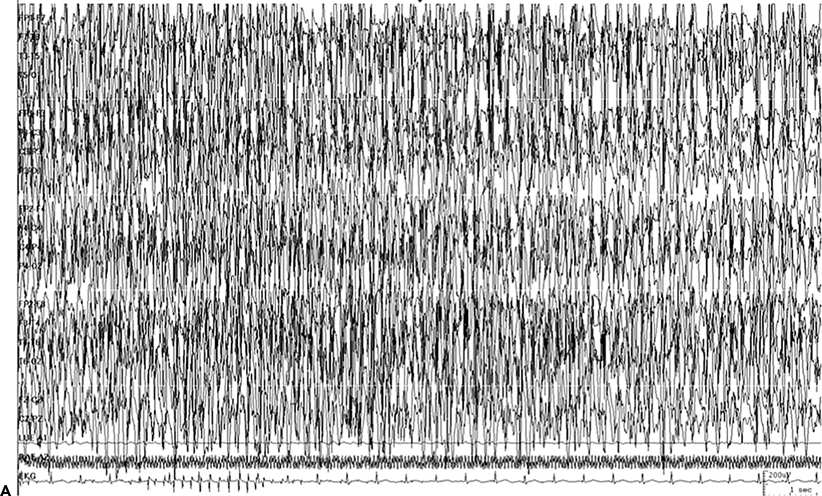
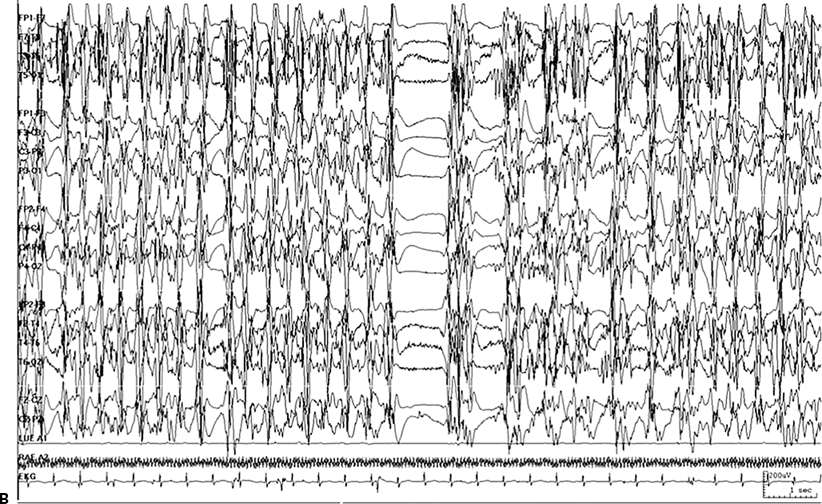
Figure 38.2. Same patient, different seizure. A: Continuous ictal discharge. B: Continuous ictal discharges punctuated by flat periods.
TRENDS IN PATIENTS WITH STATUS EPILEPTICUS
As prolonged seizures are unlikely to spontaneously cease, the overall trend in SE has been to decrease the time duration required for diagnosis and to treat as soon as possible. Although the EFA Working Group defined SE as a seizure duration >30 minutes, the Working Group recommended treatment as soon as 10 minutes after seizure onset (5). Lowenstein et al. (6) proposed an operational definition for generalized convulsive SE in adults and older children (>5 years of age) of ≥5 minutes of either a continuous seizure or two or more discrete seizures between which there is incomplete recovery of consciousness. In the Veterans Affairs (VA) Cooperative Study (26), which compared various first-line antiepileptic drugs (AEDs), treatment for SE was initiated at 10 minutes, and the San Francisco Prehospital Treatment study used 5 minutes (27). Beran has questioned waiting even as long as 5 minutes to treat an ongoing seizure (28). Similarly, Lowenstein and Alldredge had recommended immediately proceeding to anesthesia in the special case of SE developing while in the ICU (2). Table 38.2 lists other special circumstances for which immediate treatment during the early stage or even the incipient stage of SE (see Table 38.1) is recommended (29).
Table 38.2 Special Circumstances of the Early Stage

Clinical data characterizing the duration of a typical seizure support this trend of earlier treatment. A typical clinical seizure rarely lasts as long as 5 minutes. A typical generalized tonic–clonic seizure lasts 31 to 51 seconds, with a postictal phase of a few seconds to 4 minutes (30). In an inpatient study, mean seizure duration was 62 seconds, with a range of 16 to 108 seconds (31). In complex partial seizures in children, the typical duration was 97 seconds (32). In a prospective study of new-onset seizures in children, the frequency distribution of seizure duration was best described as the sum of two groups: one with a mean of 3.6 minutes (76% of cases) and the other with a mean of 31 minutes (24% of cases); if the seizure duration was 5 to 10 minutes, it was unlikely to cease spontaneously within the next few minutes (33).
The trend for the prompt treatment of SE during the early stage is supported by clinical and experimental studies characterizing the treatment of SE. In the prospective VA Cooperative Study, the first-line treatment of shorter-duration overt generalized SE was more successful than the first-line treatment of the more prolonged subtle SE, independent of treatment arm. In addition, in post hoc analysis, it was demonstrated that when the first-line AEDs failed, there was only a 5.3% response to a third AED (22). Although the response rate to treatment with a third AED was higher (58%) in a retrospective study of 83 episodes of SE in 74 patients (34), it has been posited that this difference reflects earlier treatment.
A time-dependent efficacy of treatment has also been observed in experimental models of SE. Following induction of SE with the combination of lithium and the cholinergic agonist pilocarpine, diazepam was effective in controlling SE shortly after onset but was effective in only 17% of rats in the late electrographic stages of SE (35). This finding was later confirmed in a second model of SE for both diazepam and phenytoin (36). This decrease in the benzodiazepine response occurs rapidly after the onset of SE and in young animals with ages corresponding to a human toddler (37–39). Diazepam and the other benzodiazepines enhance the function of a subset of benzodiazepine-sensitive GABAA receptors. Several studies (40–43) have demonstrated that the surface expression of these GABAA receptors declines during SE and have postulated that this reduction in surface expression, which is the result of activity-dependent, subunit-specific trafficking of GABAA receptors, partially accounts for the time-dependent efficacy of the benzodiazepines.
PATHOPHYSIOLOGY
Mechanistically, SE occurs when factors that “normally” terminate seizures fail allowing the seizure to persist and self-propagate (5,44). What are these pathophysiologic mechanisms? In simplistic terms, SE results from decreased cerebral inhibition, excessive cerebral excitation, or a combination of both. A rapid modification in the properties of GABAA receptors (37,45) and excitatory amino acid receptors (46,47) through mechanisms such as altered receptor trafficking (see above) likely contributes to the reduction in inhibition and increase in excitation.
Excessive excitation itself may cause neuronal injury and cell death, referred to as excitotoxic injury. This has been demonstrated in experimental models, such as in kainic acid–induced limbic seizures (48), but its occurrence in humans had been questioned. An outbreak of domoic acid poisoning, an excitotoxic agent, with acute symptoms, including SE, was associated with neuronal loss and astrocytosis that was greatest in the hippocampus and amygdala; this is similar to the seizures induced by kainic acid (49,50). A survivor developed epilepsy and, after death, autopsy revealed hippocampal sclerosis (51).
Prolonged seizures in anesthetized baboons cause irreversible neuronal injury (52,53). Lothman outlined the alterations in systemic and brain metabolism occurring with prolonged SE (54): decreased brain oxygen tension, mismatch between the sustained increase in oxygen and glucose utilization and a fall in cerebral blood flow, and depletion of brain glucose and oxygen. In the incipient or early stages of SE, brain compensatory mechanisms may protect against neuronal injury. However at some point, the ability to compensate for neuronal injury is exhausted, and the risk of neuronal injury increases. This point defines the transition stage from early to late (established) SE. During all stages, the ability to compensate requires adequate airway and good breathing, circulation, and cerebral blood flow.
EPIDEMIOLOGY OF STATUS EPILEPTICUS
There have been two large, population-based studies in the United States—one performed in Richmond, Virginia (55) and the other in Rochester, Minnesota (56). These two studies estimate that 60,000 to 150,000 episodes of SE occur per year in the United States resulting in approximately 55,000 deaths per year. Overall, SE accounts for 1% to 8% of hospital admissions for epilepsy. Between 4% and 16% of patients with epilepsy will have at least one episode of SE, with one-third of the cases occurring as the presenting symptom in patients with a first unprovoked seizure, one-third in patients with established epilepsy, and one-third in those with no history of epilepsy (57).
The incidence has varied by location: the rate in the study performed in Richmond was 41/100,000 (55) and the rate in the Rochester study was 18/100,000 (56). Lower rates were observed in a study performed in California (58), where the overall rate of generalized convulsive SE was 6.2/100,000, and in European studies, where the incidence rate was 9.9/100,000 in Switzerland (59), 15.8/100,000 in Germany (60), and 13.1/100,000 in Bologna (61).
A consistent finding across studies is that higher incidence rates occur at the extremes of life. For example, in the California study, the incidence rate for children <5 years of age was 7.5/100,000 and the incidence rate for the elderly was 22.3/100,000. In children, SE is most common in the very young, especially those <2 years of age (62). In the community- based prospective North London Status Epilepticus in Childhood Surveillance Study (NLSTEPSS), the incidence of SE during childhood was from 17 to 23/100,000/year (63). For children with epilepsy, SE typically occurs within 2 years of the onset of epilepsy (64), and recurrent SE is more likely with an underlying neurologic disorder (65).
ETIOLOGY OF STATUS EPILEPTICUS
As previously noted, SE is not a disease but the end result of multiple causes. These causes may be acute or remote to the episode. There may be a remote cause for the epilepsy with a new acute precipitant for the episode of SE. SE may be observed as part of a progressive encephalopathy resulting from a genetic disorder. In some cases, the cause of the SE may remain unknown (66).
In several studies of adult SE, trauma, tumor, and vascular disease were the most frequently identified causes, although idiopathic and unknown causes were also common (67–70). Etiology also differs among centers and by ages. In San Francisco, noncompliance with AEDs and alcohol withdrawal were the two most common etiologies (Table 38.3) (67,70), whereas cerebrovascular damage was the most common etiology in Richmond (71).
Table 38.3 Etiology in the San Francisco Studies: Changes Over Time
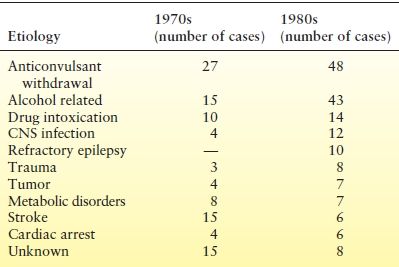
CNS, central nervous system.
From refs Lowenstein DH, Alldredge BK. Status epilepticus at an urban public hospital in the 1980s. Neurology. 1993;43:483–488; Aminoff MJ, Simon RP. Status epilepticus. Causes, clinical features and consequences in 98 patients. Am J Med. 1980;69:657–666.
For children in North London, the age-adjusted incidence for acute symptomatic SE was 16.9% in those <1 year of age, 2.5% in those 1 to 4 years, and 0.1% in those 5 to 15 years of age. The incidence of an acute on remote (remote symptomatic with an acute precipitant) was 6%, 5.3%, and 0.7%, respectively. A prolonged febrile seizure occurred in 4.1/100,000, acute symptomatic causes in 2.2/100,000, remote symptomatic in 2.3/100,000, acute on remote in 2.1/100,000, idiopathic in 1.4/100,000, cryptogenic in 0.2/100,000, and unclassified in 1/100,000 (63).
As the Richmond study included adults and children, the etiologies in these two groups can be directly compared (Table 38.4). In adults, cerebrovascular disease was the most common etiology, occurring in 25.2%, versus only 3.3% in children, whereas in children, fever or infection was the most common cause, occurring in 35.7% versus only 4.6% in adults. Medication change was a major cause in both adults and children—20% in children versus 19% in adults (71). The incidence of tumors was higher in older studies (68,69).
Table 38.4 Comparison of Etiology in Children and Adults in the Richmond Study
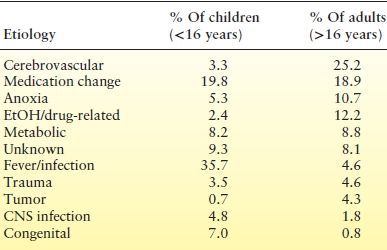
CNS, central nervous system.
From ref DeLorenzo RJ, Towne AR, Pellock JM, et al. Status epilepticus in children, adults, and the elderly. Epilepsia. 1992;33(suppl 4):S15–S25.
PROGNOSIS OF PATIENT WITH STATUS EPILEPTICUS
Although many people may survive an episode of SE, even refractory SE, with no or limited untoward effects (72,73), SE can be life threatening and has been associated with long-term neurologic sequelae. The prognosis of SE depends partially on etiology (74), duration (2,75), and age (62). Recently, concern has been raised that prognosis may be affected by the use of anesthetic agents and the need for intubation and mechanical ventilation (76).
The mortality rate in modern, general SE series ranges from 4% (77) to 37% (78) and is higher with an acute precipitant (78). An acute precipitant is more likely when there is no prior history of epilepsy (78,79), but may also be responsible for death in persons with known epilepsy with SE. In one series, 63% of patients survived, 28.6% died from the underlying cause, 6.6% died from other causes, and 1.8% died from the SE itself (78). In a retrospective study of 74 patients (80), the mortality rate was 21% (14/85) and was higher with acute symptomatic seizures and older ages. Interestingly, in a study limited only to patients with de novo SE (81), SE occurring in patients already hospitalized, the mortality was very high—61% (25/41).
Short-term versus long-term mortality was compared using data from the Rochester study (82–84): Mortality was 19% (38/201) within the first 30 days, but cumulative mortality was 43% over 10 years (82). The long-term mortality risk increased with an SE duration >24 hours, acute symptomatic etiology, and myoclonic SE (82).
In the Richmond study, the mortality rate was 32% when the duration was >60 minutes versus only 2.7% when the duration was 30 to 59 minutes (85). Other factors associated with a high mortality rate in the Richmond study included anoxia and older age, whereas a low mortality rate was associated with alcohol and AED withdrawal (85).
In the Netherlands study (17), prognosis of patients with generalized convulsive SE was related to treatment adequacy. A favorable outcome occurred in 263 of 346 patients (76%), with outcome related to cause, duration >4 hours, more than one medical complication, and quality of care. In order to analyze the treatment effects, therapy was classified as insufficient when the wrong AED dose or route was used, if an unnecessary delay occurred, if mechanical ventilation was not used despite respiratory insufficiency or medical complications, or if neuromuscular paralysis was used without EEG monitoring (in order to detect seizure activity). The most common reason for classifying therapy as insufficient was an inadequate AED dose. In the patients with a favorable outcome (n = 263), therapy was classified as good or sufficient in 85.6% and considered insufficient in only 10.3%; in those with sequelae (n = 45), therapy was inadequate in 22.2%. When the morbidity was from SE itself, insufficient therapy occurred in 50% of patients. With the occurrence of death (n = 38), therapy was sufficient in 44.7% of patients, and in cases of death due to SE itself, therapy was considered insufficient in 62% of patients (17).
The mortality rate in pediatric SE ranges from 3% to 11% and is also related to etiology and age (77,86–92). In one study, the mortality was 4%, occurring only with acute symptomatic or progressive symptomatic etiologies (86). In NLSTEPSS, the overall mortality was 3%. In the Richmond study, for children ranging in age from 0 months to 16 years (n = 598), the overall mortality rate was 6.2% (37 of 598). The highest rates occurred during the first 6 months of life (24%; 18 of 75) and between 6 and 12 months of age (9.25%; 5 of 54) with a low rate (2.98%; 4 of 469) in children older than 1 year (93). The difference likely reflects a higher incidence of symptomatic SE in the youngest children. With respect to morbidity following SE in children, a minority of children will have new-onset motor dysfunction and loss of previously obtained milestones (77,92). A Canadian study of SE reported 34% of 40 children with an SE duration of 30 to 720 minutes had subsequent neurodevelopmental deterioration (94). Even in children with febrile SE, speech deficits have been reported (95).
An increase in morbidity and mortality has also been reported with NCSE, which is related to SE duration (36 hours to >72 hours) (96). However, this increased morbidity with NCSE is controversial (97–99). Following cardiopulmonary resuscitation, SE, status myoclonus, and myoclonic SE are predictive of a poor outcome (100). On EEG, burst suppression (101) and PEDs are predictive of a poor outcome (102), whereas a normal EEG is associated with a good prognosis (103).
MANAGEMENT OF STATUS EPILEPTICUS
The initial management of patients with SE begins with the ABCs—airway, breathing, and circulation (Table 38.5). Diagnostic studies are then selected, depending on a patient’s history and physical examination (not all studies need to be obtained for every patient). Serum glucose should be checked immediately with bedside glucose testing to rapidly diagnose hypoglycemia. A complete blood count may be helpful for diagnosing infection, although leukocytosis may occur with SE. Electrolytes, calcium, phosphorous, and magnesium values may also be helpful. Lumbar puncture (LP) should be considered in the febrile patient, although CSF pleocytosis may occur without infection, presumably due to a breakdown in the blood–brain barrier (104). In one study, the highest CSF white blood cell count from SE alone (no acute insult) was 28 × 106/L (105). If there is concern about increased intracranial pressure or a structural lesion, LP can be deferred until neuroimaging is performed. If there is evidence of infection, antibiotics can be administered prior to LP. In those taking an AED, levels should be obtained as low AED levels may contribute to the development of SE in both adults and children (106,107). A practice parameter on the diagnostic assessment of the child with SE has been produced (108). When done, electrolytes or glucose were abnormal in 6%, blood cultures were abnormal in 2.5%, a CNS infection was found in 12.5%, an ingestion was found in 3.6%, an inborn error of metabolism was found in 4.2%, and AED levels were low in 32%.
Table 38.5 Immediate Management of Status Epilepticus

ECG, electrocardiogram; CBC, complete blood count; AED, antiepileptic drug; CAT, computerized axial tomography; MRI, magnetic resonance imaging; EEG, electroencephalography.
Neuroimaging options include cranial computed axial tomography (CAT) scan and magnetic resonance imaging (MRI). CAT scans are readily available on an emergency basis and should identify all disorders demanding immediate intervention, such as tumor or hydrocephalus, but may not show the early phases of infarction. CAT scan and MRI may detect focal changes, which may be transient (109), secondary to a focal seizure (suggesting the origin of the focus), with MRI the more sensitive technique. Although these lesions may mimic those of ischemic stroke, they are reported to cross vascular territories (110). Changes in diffusion-weighted images and the apparent diffusion coefficient suggest both cytotoxic and vasogenic edema (111). Progressive changes also occur, such as hippocampal atrophy and sclerosis, or global atrophy (112,113). In the FEBSTAT study, a prospective study of children between 1 month and 5 years of age who have experienced an episode of febrile SE, 9% (17/191) were found to have increased T2 signal in the hippocampus, maximum in Sommer sector, when imaging was completed within 72 hours of presentation, and another approximately 2.5% (5/191) had an equivocal finding. None of the 96 children with a first simple febrile seizure who made up the control group were found to have a similar increase in signal (114). Repeat imaging in this cohort obtained approximately 1 year later demonstrated retarded hippocampal growth after febrile SE and, in several of the children with acute findings, the development of imaging characteristics of hippocampal sclerosis (115). The North London Group found in a cohort of 80 children that 20% to 30% of the children (0.18 to 15.5 years) had evidence of hippocampal volume loss on MR imaging of the brain 1 month following an episode of convulsive SE of any etiology (116). In a fatal case of unexplained SE, high-signal lesions in the mesial temporal lobes and hippocampal neuronal loss were reported (117). In general, neuroimaging should be performed in all patients with new-onset SE, especially if there is no prior history of epilepsy.
Intoxication with certain agents, particularly theophylline (79,81,118) and isoniazid (INH) (119), which may involve acidosis (120) and is treated with pyridoxine (vitamin B6) (121), may predispose individuals to generalized convulsive SE or NCSE. Immunosuppressants such as cyclosporine (122) or tacrolimus and ifosfamide (123) may predispose individuals to NCSE, which may also occur when phenytoin and carbamazepine are used in patients with idiopathic generalized epilepsy (124); lithium (125), tiagabine (126), and amoxapine (127) may also be implicated. Fatal SE has occurred with flumazenil; therefore, caution should be exercised in patients with a history of seizures and chronic benzodiazepine use or when a mixed overdose is suspected (128).
An EEG is not immediately needed for treatment. Indications for emergency EEG include unexplained altered awareness (to exclude NCSE) (Fig. 38.3), the use of neuromuscular paralysis in a patient with SE, high-dose suppressive therapy for refractory SE, and no return to baseline or improvement in mental status following control of overt convulsive movements (to exclude ongoing subtle SE) (129). NCSE occurs in 14% of adults (130) and 26% of children (131) in whom generalized convulsive SE has been controlled after treatment. In a retrospective study of children who experienced an episode of convulsive SE and subsequently underwent continuous EEG monitoring in an intensive care setting, a third were discovered to have electrographic seizures (132). In another study, NCSE was detected in 8% of all comatose patients (101). Therefore, the EEG should be used when the diagnosis is in doubt, especially in patients with pseudoseizures.
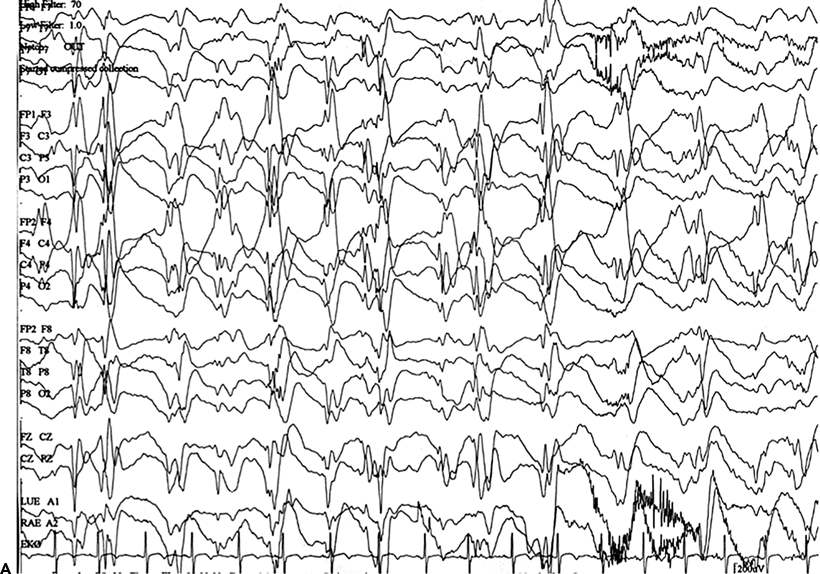
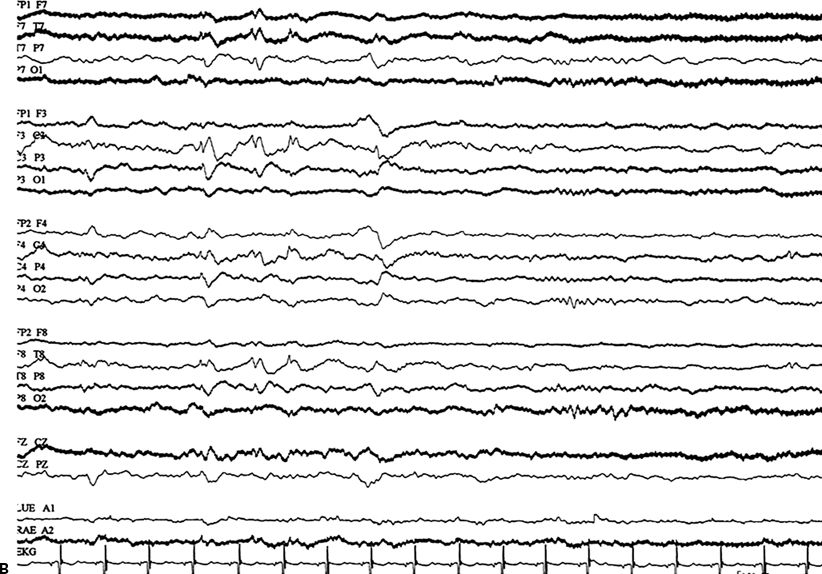
Figure 23.3. A:Nonconvulsive SE: continuous ictal discharges, slow spike and wave, with altered awareness. B:Nonconvulsive SE: electroencephalogram after lorazepam, now with improved awareness.
ANTIEPILEPTIC DRUG THERAPY FOR STATUS EPILEPTICUS
Treatment should be aimed at controlling SE as soon as possible, particularly before brain compensatory mechanisms fail. Despite adequate oxygenation and ventilation, such failure has been reported within 30 to 60 minutes in experimental SE (54) and within 30 to 45 minutes in humans (2). Systemic and metabolic changes occur early, with increases in blood pressure, lactate, and glucose levels. Both respiratory and metabolic acidosis may develop, although the former is more common (133). Initially, brain parenchymal oxygenation, lactate, glucose, and oxygen utilization remain stable, cerebral blood flow increases, but cerebral glucose slightly decreases. In later stages, blood pressure may be normal or decrease slightly, glucose may decrease, and hyperthermia and respiratory compromise may occur, leading to hypoxia and hypercarbia. Decreases in brain parenchymal oxygenation, cerebral blood flow, and brain glucose all contribute to an energy mismatch (54). Neuron-specific enolase, a marker of brain injury, is elevated in the serum following both convulsive and NCSE (134,135).
Neuronal injury may occur in the absence of metabolic derangement. In paralyzed and ventilated baboons given bicuculline, a GABAA receptor competitive antagonist, to induce electrographic SE (52,53), neuronal loss was observed in the neocortex and hippocampus. Brain lesions following flurothyl-induced SE in the paralyzed and well-oxygenated rat include hypermetabolic infarction of the substantia nigra (136). In humans, neuronal loss was seen following SE in three patients without hypotension, hypoxemia, hypoglycemia, or hyperthermia (137).
Most of the AEDs used to treat SE have the potential for respiratory and cardiac depression, especially when administered by a loading dose (138) or when an excessive dose is used (139). Therefore, protecting the airway, controlling ventilation, and monitoring cardiac and hemodynamic function are mandatory. Intravenous (IV) administration is the preferred route for the treatment of SE, especially in the inpatient setting, but if IV access is difficult, intramuscular (IM), rectal, or intranasal routes have been used.
Primary AEDs for SE
The benzodiazepines (lorazepam, diazepam, midazolam), phenytoin or its prodrug form fosphenytoin, and phenobarbital are the current drugs of first choice for the initial therapy in patients with SE (Table 38.6). However, some advocate the early use of the secondary agents (e.g., valproic acid, levetiracetam, anesthetics).
Table 38.6 First-Line (Emergent Initial Therapy) Intravenous Antiepileptic Drugs
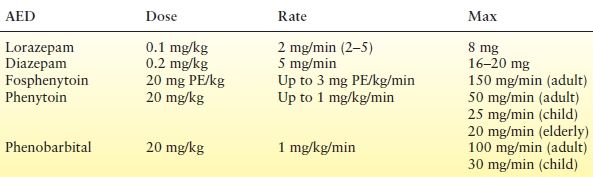
Treatment of SE is typically initiated with a benzodiazepine. Diazepam was rapidly applied to the treatment of SE soon after its discovery and has remained a mainstay of treatment. Theoretically, compared to lorazepam, diazepam should have a more rapid onset of action because of greater lipid solubility (138). However, this high lipid solubility results in a rapid redistribution to inactive tissues, such as fat, which can result in high rates of seizure recurrence (140,141).
Lorazepam has been used in both adults and children (142,143). Although expected to have a slower time to onset than diazepam, the times of onset of lorazepam 4 mg and diazepam 10 mg in the treatment of SE in adults were similar (144). In that double-blind study, SE was controlled in 89% of episodes with lorazepam versus 76% with diazepam, with similar rates of adverse events (144). Because of a smaller volume of distribution, lorazepam should have a longer anticonvulsant activity and, thus, a lower rate of seizure recurrence than diazepam (145,146). In a recently completed prospective randomized multicenter trial, lorazepam 0.1 mg/kg IV and diazepam 0.2 mg/kg IV were equally effective in terminating convulsive SE in children (3 months to younger than 18 years) in the emergency department. Interestingly, at these doses, there were no differences in the rate of recurrence up to 4 hours (147).
Midazolam, due to its water solubility, may be administered intranasally or intramuscularly if there is no IV access, and it has been associated with less sedation and respiratory depression (148). The efficacy of intramuscular midazolam in the out-of-hospital treatment of convulsive SE in children and adults was prospectively compared to intravenous lorazepam in the double-blind, randomized RAMPART trial (149). Upon emergency department arrival, seizures had stopped in 73.4% of the subjects in the midazolam group and 63.4% in the lorazepam group. Seizure recurrence was low in both groups, and there was no difference in adverse events. Although designed as a noninferiority study, the results do support statistical superiority of intramuscular midazolam in the prehospital treatment of convulsive SE.
Phenytoin may be administered by an IV loading dose in normal saline (it precipitates with dextrose) at 20 mg/kg (15 mg/kg in the elderly), which rapidly achieves a therapeutic level without respiratory depression or sedation and can also provide maintenance therapy (150–152). This lack of sedation is important for monitoring mental status, such as in patients with head trauma. The infusion rate should be no faster than 1 mg/kg/min in a child (not to exceed 25 mg/minute), 50 mg/minute in an adult, and 20 mg/minute in the elderly. Pulse and blood pressure should be monitored. If hypotension develops, the infusion rate should be decreased. In adults, a therapeutic level should be maintained for up to 24 hours after a loading dose has been administered (153), but may not last as long in children (153). A level obtained 2 hours after loading may help guide the timing of maintenance therapy with phenytoin (153).
IV phenytoin has an alkaline pH and contains solvents that can cause vascular irritation, cardiac depression, and hypotension. The purple glove syndrome, consisting of distal limb edema, discoloration, and pain, may occur following IV phenytoin infiltration; treatment may require fasciotomies and amputation. In one series, purple glove syndrome occurred in nine of 152 patients (154); in a prospective series, it occurred in only three of 179 patients (155). The syndrome has also been reported following oral dosing in a child (156).
The phosphate ester prodrug of phenytoin, fosphenytoin, is dosed as phenytoin equivalents (PE) at 20 mg PE/kg. It can be administered in a dextrose solution. Fosphenytoin is water soluble and may be given by the IM route, with paresthesias and injection site pruritus as possible adverse effects. Bioavailability is 100% compared to phenytoin, and the conversion half-life is 7 to 15 minutes (157). Fosphenytoin is rapidly converted to phenytoin by serum and tissue alkaline phosphatases (158). It may be difficult to maintain therapeutic levels in infants, and additional doses may be required (159); subtherapeutic free phenytoin levels also occur in older children (160). A 2-hour phenytoin level is suggested to ensure conversion (160). Side effects are more likely in patients with hypoalbuminemia, renal failure, or hepatic failure and in the elderly, because of the presence of higher free phenytoin levels. In these patients, the infusion rate should be decreased by 25% to 50% (157). The only advantage of IV phenytoin over IV fosphenytoin is significantly lower cost.
Phenobarbital has been used to treat SE in all age groups. Respiratory depression and sedation occur. Caution is advised, especially when phenobarbital is administered in combination with other sedative AEDs (such as benzodiazepines). In a randomized trial of diazepam and phenytoin versus phenobarbital (10 mg/kg IV), phenobarbital resulted in a shorter median seizure time (5 minutes vs. 9 minutes) and response latency (5.5 minutes vs. 15 minutes), with a similar incidence of intubation, hypotension, and arrhythmia (161). The loading dose for phenobarbital is 15 to 20 mg/kg, administered at a rate no higher than 100 mg/minute in older children and adults and 20 mg/kg in neonates and infants (160).
The landmark VA Cooperative Study (26) compared the efficacy of various first-line agents—lorazepam (0.1 mg/kg), phenobarbital (15 mg/kg), diazepam (0.15 mg/kg) plus phenytoin (18 mg/kg), and phenytoin alone (18 mg/kg)—in the treatment of SE with successful treatment defined as control of the seizure within 20 minutes (27). Treatment efficacy of overt generalized convulsive SE was similar with lorazepam (65%), phenobarbital (58%), and diazepam plus phenytoin (56%), whereas phenytoin alone was associated with lower efficacy (44%). This result may be related to a 4.7-minute infusion time with lorazepam versus 33 minutes with phenytoin alone.
Second-Line Agents for Status Epilepticus
Sodium valproate (VPA) has been available in IV form since 1995 (Depacon, Abbott Laboratories, North Chicago, IL) (162). Although it is not yet approved by the U.S. Food and Drug Administration (US FDA) for the treatment of SE, doses of 15 to 33 mg/kg have been administered safely in adults (163–172) at a rate of 20 to 50 mg/minute (164). In a review of 13 elderly patients with SE and hypotension, a mean loading dose of 25 mg/kg at 35 mg/minute was associated with no change in blood pressure (165). In one study, an infusion rate of 3 mg/kg/min was associated with hypotension in 2 of 72 patients (168).
One study has suggested that VPA may be an appropriate first-line medication under narrow conditions (173). Two studies have evaluated IV phenytoin versus IV VPA as first-line therapy for SE. The first used IV VPA 30 mg/kg over 15 minutes or IV PHT 18 mg/kg at a rate of 50 mg/minute; if SE continued, the other AED was used (174). Used as initial therapy, SE was controlled in 66% with VPA versus 42% with PHT; in the refractory patients, VPA was effective in 79% versus 25% with PHT. The side effects were similar. In the second study (175), there was equal efficacy (88% for both) with side effects of 12% with PHT versus none with VPA. In diazepam-resistant SE, 31/41 (76%) episodes of SE were controlled with a VPA 25 mg/kg loading dose over 30 minutes. The probability of successful treatment with VPA was time-dependent: if the VPA was given early (within 3 hours), next-line therapy with anesthesia was required in only 5%, but when VPA was given later, anesthesia was needed in 60% (176).
In children, loading doses of 10 to 30 mg/kg have been used, with most using the higher-dose ranges; an infusion rate of 1 mg/kg/h was not associated with serious side effects (169). A 20-mg/kg loading dose should produce a serum level of 75 mg/L (170). Valproate is safe in adults and children (163,168,172). One study reported on 48 IV VPA doses, mean 22 mg/kg (range 7.5 to 41.5 mg/kg) with a mean infusion rate of 5 mg/minute, with only 1 adverse event, burning at the infusion site (177). Hypotension occurred in one child at an infusion rate of 30 mg/kg/h (0.5 mg/kg/min) (171). A loading dose of 10 to 25 mg/kg over 30 minutes has been used in neonates (178).
Levetiracetam (LEV) is available in an IV preparation and has been used to control SE (179,180), typically as second-line therapy. A recent retrospective multicenter observational study identified that this agent was not uncommonly used following benzodiazepine failure (181). The pharmacokinetic profile is similar for IV and oral LEV with no difference following an oral or IV dose of 500 to 1500 mg in adults (182). LEV levels peak within 2 hours, a steady state is achieved within 2 days, and there are no significant drug interactions (183). IV LEV with a mean loading dose of 944 mg (range 250 to 1500 mg) controlled 16/18 episodes of SE following benzodiazepine failure (184). A 20 mg/kg loading dose followed by 15 mg/kg BID after 6 hours controlled SE in 82% overall and in 11/12 (92%) as first-line therapy (185). 2/50 (4%) developed thrombocytopenia. In a study comparing the effectiveness of PHT, VPA, and LEV following benzodiazepine failure, LEV 20 mg/kg was found to fail more often than VPA 20 mg/kg. In that study, LEV accounted for approximately 17% of second-line treatment failures (186). In nine children with acute seizure exacerbations or refractory SE, a loading dose of 10 to 30 mg/kg was given over 30 minutes, with a mean dose of 228 mg/kg/d. One child had an increase of seizures, and no agitation or behavioral problems occurred (187).
Lacosamide, which enhances the slow inactivation of voltage-gated sodium channels, was approved by the U.S. FDA in 2008 for adjunctive therapy for focal-onset seizures in patients 17 years of age and older. However, as it is available in an intravenous formulation, it is not surprising that it has been used for the treatment of SE with some success in both adults and children (188–192). A loading dose of 400 mg appears to be more effective than 200 mg in the treatment of refractory seizure clusters and SE (193). In a more recent retrospective study, lacosamide (median bolus dose of 400 mg) as a third-line agent administered at a median of 19 hours after SE onset following the failure of a benzodiazepine and LEV successfully terminated the status in 7 of 21 patients (194).
TREATMENT GUIDELINES FOR STATUS EPILEPTICUS
Standard treatment guidelines are needed in advance of all medical emergencies, to improve the quality of emergency care (195,196). The treatment guideline can then be analyzed and modified if the data demonstrate that the protocols are not resulting in improved care (197,198). To this end, there are a few randomized clinical trials, the EFA Working Group timetable (4), treatment surveys (199), and various societies’ treatment guidelines, that can assist in this process (200,201). A survey of the UK Intensive Care Society revealed that only 12% of the respondents used a specific protocol (202) and that first-line therapy was frequently with a benzodiazepine plus phenytoin. In a US survey of neurologists and intensivists (N = 106), 76% used lorazepam first, with 95% using phenobarbital or phenytoin if lorazepam failed (203). A survey of epileptologists was conducted to establish consensus guidelines for first-line, second-line, and third-line treatment options for epilepsy syndromes (204). A treatment of choice was determined if selected by >50% of respondents. Lorazepam was considered the treatment of choice for generalized convulsive, focal, and absence SE, with diazepam or phenytoin considered first-line treatment for generalized convulsive SE and focal SE; diazepam and sodium valproate were considered first-line treatment for absence epilepsy (204).
Prior to the VA Cooperative Study, the EFA Working Group suggested either lorazepam or diazepam as first-line therapy, but now, lorazepam is initially used by many (2,203,204). European guidelines have used either lorazepam or diazepam for first-line therapy. Most guidelines still use either phenytoin or phenobarbital if a benzodiazepine fails, but there is increasing use of either VPA or LEV. We use 0.1 mg/kg lorazepam initially for children, at a maximum dose of 4 mg when IV access is available; if IV access is not available, diazepam or lorazepam can be administered rectally or fosphenytoin or midazolam via the IM route. However, although there are theoretical reasons to support the use of lorazepam over diazepam (as discussed above), a review of randomized clinical trials in children found no evidence that treatment with IV lorazepam was better than treatment with diazepam (205), which is further supported by the results of a prospective, randomized clinical trial comparing lorazepam to diazepam in the treatment of pediatric SE (147).
The Neurocritical Care Society guideline for the evaluation and management of SE states that definitive control of SE should be obtained within 60 minutes of onset. Treatment occurs in stages, first line through fourth or fifth line. This guideline suggested the use of emergent initial therapy (first line), urgent control therapy (second line), and refractory therapy (third through fifth line) in order to emphasize the need for the emergent control of SE (201).
Treatment of Refractory Status Epilepticus
Refractory SE occurs when seizures persist despite adequate treatment with a first- and second-line agent (201). The mortality in adults with refractory SE varies from 39% to 48% (206) and in children, from 16% to 43.5% (66,207,208). Etiology is a very important determinant, with a higher mortality among acute symptomatic patients (17,34,66,78,80). In children, the data also demonstrate that etiology is related to prognosis (66).
If convulsive activity has stopped but mental status does not improve, NCSE must be excluded, which occurs in 14% of adults (130) and 25% of children (131), and in 8% of those with unexplained coma (101). An immediate EEG is performed, if available; if not available, additional empiric AED therapy must be considered.
If SE persists following the use of the second conventional AED, then continuous intravenous infusion AEDs should be administered (Table 38.7). The treatment goal is to stop SE immediately and to prevent seizure recurrence. However, it is important to recognize that continuous infusion AEDs have been linked with several untoward effects including mechanical ventilation, hypotension, and infection as well as potentially increasing the risk of death (76,209–212).
Table 38.7 Agents Used in Refractory Status Epilepticus

Pentobarbital has been the most widely used agent under these circumstances (213–220), administered at 2 to 10 mg/kg followed by a continuous infusion. Midazolam has a shorter half-life and is associated with less sedation (148,221–226). Adults with RSE with maximum midazolam infusion rates of approximately 3 mg/kg/h were found to have less withdrawal seizures and potentially lower risk of mortality at discharge compared to those in which the maximum infusion rate was lower at 0.4 mg/kg/h (227). High-dose phenobarbital is also used; it is associated with less cardiovascular depression than pentobarbital (228,229) but has a longer half-life.
Other agents used include benzodiazepines (230,231), thiopental (232), lidocaine (233–235), inhalational anesthetics such as isoflurane (Forane, Baxter Pharmaceuticals, Deerfield, IL) (235–237), and propofol (238,239). Propofol has two main advantages: a rapid onset and a short duration of action. One study with pentobarbital and propofol in adults showed equal efficacy, but propofol controlled SE in 2.6 minutes versus 123 minutes with pentobarbital (238). Another study in children with RSE reported an efficacy of 64% with propofol versus 55% with thiopental, and no side effects with propofol, with infusion rates <5 mg/kg/h (240,241). Propofol may cause metabolic acidosis with prolonged use in children (242,243). In adults, deaths have occurred with high propofol infusion rates (244), which is known as propofol infusion syndrome (245). Even in an adult study that showed equal efficacy for seizure control, a 57% mortality rate was reported with propofol versus only 17% with midazolam (221). Therefore, propofol should be used with caution, especially in children and ideally for a short time only, and the infusion rate should not exceed 67 μg/kg/min (246). Immediate control can be achieved and then another agent used if long-term suppression is needed.
The N-methyl-d-aspartate receptor antagonist ketamine may also have a role in the treatment of RSE (247–252). A recent multicenter retrospective review of 58 subjects from 10 academic medical centers treated with ketamine found that ketamine was potentially successful in up to a third of the episodes of SE. Ketamine was not successful when used after a failure of seven or more drugs or when introduced at least 8 days after SE onset. In those in which ketamine was deemed to contribute to the control of SE, the dosing was >0.9 mg/kg/h. Overall, ketamine was found to be well tolerated with many of the untoward effects that occurred during the ketamine dosing being attributed to the concurrent medications (253).
Chlormethiazole (254), etomidate (255), and clonazepam (256) are used in Europe; paraldehyde (257) and chloral hydrate (258) may be administered rectally, although paraldehyde is no longer available in the United States. Hypothermia (259–261), vagal nerve stimulation (262), and surgery (263) have also been used. The ketogenic diet may also have a role, especially in children diagnosed with febrile infection–related epilepsy syndrome (264–266).
To date, no prospective study has been conducted in patients with refractory SE. In a systematic review of refractory SE treatment with pentobarbital, propofol, or midazolam (267), pentobarbital was associated with better seizure control than the other two agents. In the UK survey (N = 408), if first-line treatment failed, 142 (35%) of the respondents used a benzodiazepine infusion and 130 (32%) used a general anesthetic. If seizures continued, 333 (82%) used thiopentone and 56 (14%) used propofol (202). Based on the consensus guidelines, the drug of choice for “therapeutic coma” in patients with generalized convulsive SE and focal SE was pentobarbital, and first-line agents were midazolam and propofol; for absence seizures, pentobarbital was the drug of choice, with no other first-line options, and midazolam was considered second-line therapy (204). In the US survey, when generalized convulsive SE was refractory to two AEDs, 43% of respondents used phenobarbital, 16% used valproate, and 19% gave one of three agents (pentobarbital, midazolam, or propofol) by continuous infusion (203).
Whether clinical seizures alone or both clinical and electrographic seizures need complete control is controversial (201,268,269). In this situation, many clinicians use continuous infusion AEDs titrated to a burst suppression pattern on EEG, aiming for complete control of both the clinical and electrographic seizures. Some aim only for control of clinical seizures (without EEG monitoring). In the US survey, the titration goal with a continuous infusion was burst suppression in 56% of respondents versus elimination of seizures in 41% (203), whereas in a European survey, up to 70% titrated the EEG to a burst suppression pattern (270). However, the outcome is not related to the extent of EEG burst suppression and is more dependent on etiology (271).
Even if a burst suppression pattern is the goal, the degree of suppression needed is unclear. We have used a burst suppression pattern as the clinical end point, aiming for an interburst interval of at least 5 seconds in duration (66,272). In an analysis of the depth of EEG suppression with barbiturate anesthetics (pentobarbital or thiopental) in adults, persistent seizure control was better with electrocerebral inactivity on EEG (17 of 20) versus a burst suppression pattern (6 of 12 patients) (273,274). Using a midazolam infusion to eliminate all clinical and electrographic seizures and reaching burst suppression only if needed, acute treatment failure occurred in 18% of episodes, breakthrough seizures in 56%, posttreatment seizures in 68%, and treatment failure in 18% (225). In the systematic review, breakthrough seizures occurred less frequently with titration to EEG background suppression (53%) versus titration to seizure suppression only (4%). However, hypotension occurred more often with titration to background suppression (267).
Prolonged continuous infusion AEDs can be used (66,275), usually with various AED combinations. A continuous infusion AED is used initially for a short time (12 to 24 hours); the infusion is then tapered, and if SE recurs, the sequence restarts (55,66,272). Mirski et al. (276) recommended prolonged therapy with a potentially good prognosis: a healthy patient (no premorbid illness), a self-limited disease, and with neuroimaging not indicating a poor prognosis. Bramstedt et al. (277) recommended ethically withholding suppressive therapy if only expected to sustain organic life. We have treated children for prolonged periods of up to 146 days (66,275), and a 26-year-old with encephalitis was treated for 11 months (278). In our experience with children, no survivor of acute symptomatic refractory SE (n = 7) returned to baseline, and all subsequently developed refractory epilepsy; seizure recurrence was reported upon drug tapering in two children and within 1 to 16 months in the other five (225). In our entire group with refractory SE, 32% returned to baseline (66), and in the adult systematic review, only 29% (48 of 164 patients) returned to baseline (267).
Infectious or inflammatory disorders may predispose to refractory SE. In one series, 7/22 had “presumed encephalitis” (66,275). Kramer also reported severe RSE from “presumed encephalitis” (279). Holtkamp reported encephalitis as a predictor for RSE (280) and referred to this as a “malignant variant” (281). Wilder-Smith defined the syndrome of new-onset refractory status epilepticus (282). Characteristic features include female gender, young age, previous good health, CSF pleocytosis, antecedent febrile illness, and prolonged treatment. We reported complete seizure control during suppression in 5/7 in this group, with then a seizure recurrence and the development of refractory epilepsy ranging from 1 to 16 months later (275). The occurrence of this latent period raises the question if neuroprotective, antiepileptic, or even immunomodulatory agents might be helpful in this situation. Although the RSE that accompanies so-called febrile infection–related epilepsy syndrome (FIRES) has been attributed to acute neuroinflammation (283,284), the absence of a latent period has led others to hypothesis that FIRES is best conceptualized as chronic neocortical epilepsy with explosive onset (285).
Several specific repetitive disorders have been reported in SE: Stimulus-induced rhythmic, periodic, or ictal discharges (286) and cycling seizures (287) have been reported. Acute encephalitis with refractory, repetitive partial seizures has been described in children with encephalitis (288,289). These have an abrupt onset in the setting of a fever following an antecedent infection; are brief, focal seizures; occur with an escalating frequency; and are resistant to standard anticonvulsants and require high-dose suppressive therapy for control.
Prehospital Treatment
The advent of intrarectally administered AEDS permits the premonitory or early stage to be treated prior to hospital arrival (290–292), although other routes of administration are also used. The prospective San Francisco Prehospital Treatment study (N = 205) found that lorazepam was potentially more effective than was diazepam in terminating SE out of hospital (59% response with lorazepam vs. 43% response with diazepam and 21% response with placebo; P = 0.001) (27). In a retrospective study of 38 children with generalized convulsive SE, use of prehospital diazepam (0.6 mg/kg rectally) was associated with a shorter seizure duration (32 minutes vs. 60 minutes) and a reduced likelihood of seizure recurrence in the emergency department (58% vs. 85%), with no difference with respect to intubation (293). Rectal diazepam can be administered at home for the treatment of SE or serial seizures; the maximum dose is 20 mg. A rectal gel preparation, Diastat (Xcel Pharmaceuticals, San Diego, CA), is available, which is easier to administer (294–296). Although only approved by the US FDA for the treatment of selected, refractory, patients on stable regimens of AEDs for the treatment of seizure exacerbation, Diastat is used as a therapeutic remedy for SE at home; IV benzodiazepines are still preferable for the treatment of inpatients. Lorazepam can be administered sublingually (297), and midazolam can be given by intranasal or buccal mucosa routes (298), with rapid buccal absorption documented by serum levels and EEG beta activity (299). The efficacy of intranasal midazolam (0.2 mg/kg) is equivalent to that of IV diazepam (0.3 mg/kg) for the treatment of prolonged febrile seizures (300), and buccal midazolam (10 mg) and rectal diazepam (10 mg) have shown equal efficacy for seizures >5 minutes (298). As previously noted, the RAMPART study demonstrated the utility of intramuscular midazolam in the prehospital treatment of convulsive SE (149). Paraldehyde is included in a UK pediatric treatment protocol (196), but as previously noted, it is no longer available in the United States.
Emergency Department or Inpatient Treatment
Lorazepam 0.1 mg/kg should be administered initially. A suggested treatment sequence follows, as outlined in Table 38.8 (301–303).
Table 38.8 A Suggested Timetable for the Treatment of Status Epilepticus

aMay consider the use of continuous intravenous infusion AEDs after failure of the first two AEDs [RSE, Neurocritical Care Society Guideline (201)].
AED, antiepileptic drug.
Modified from refs Gastaut H. Classification of status epilepticus. Adv Neurol. 1983;34:15–35; Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338:970–976; Riviello Jr JJ, Claassen J, LaRoche SM, et al. Treatment of status epilepticus: an international survey of experts. Neurocrit Care. 2012;18:193–200; Brophy GM, Bell R, Claassen J, et al.; and the Neurocritical Care Society Status Epilepticus Guideline Writing Committee. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care. 2012;17:3–23.
References
1. Gastaut H. Classification of status epilepticus. Adv Neurol. 1983;34: 15–35.
2. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338: 970–976.
3. Delgado-Escueta AV, Wasterlain C, Treiman DM, et al. Current concepts in neurology: management of status epilepticus. N Engl J Med. 1982;306:1337–1340.
4. Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America’s Working Group on Status Epilepticus. JAMA. 1993;270:854–859.
5. Lowenstein DH, Bleck T, Macdonald RL. It’s time to revise the definition of status epilepticus. Epilepsia. 1999;40:120–122.
6. Berg AT, Berkovic SF, Brodie MJ
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


