9 Surgical Treatment of Chordomas and Chondrosarcomas Chordomas and chondrosarcomas are osteocartilaginous tumors that typically involve the skull base in a parasellar location. These tumors, often grouped together because of their similar osseous origins and location, are distinct in embryology, pathology, biological behavior, clinical features, and response to therapy. Chordomas arise from remnants of the embryonal notochord and are frequently located in the central skull base. Most grow slowly initially, but eventually their growth accelerates. They are locally invasive, and invasion, combined with rapid growth, often proves fatal. Chondrosarcomas originate from the chondral elements of bone. In the skull, they are often located in the paramedian sphenoid and the clival basiocciput. Most chondrosarcomas demonstrate slow, locally invasive growth. Many chondrosarcomas do not affect patient function or survival for decades. Chordomas and chondrosarcomas together account for ~0.2% of all intracranial tumors and 6% of all primary skull base tumors.1,2 Although chordoma is the most common extradural primary skull base tumor, its absolute incidence is low (<0.1 per 100,000 persons per year, or ~0.15% of all intracranial tumors).3 Chordomas can occur at any age, but they peak in the fourth or fifth decade of life, with a median age at diagnosis of 46 years. Fewer than 5% arise in children.4,5 There is no gender preponderance2,6 or known association with exposure to radiation or environmental carcinogens. Chordomas occur in isolation and are not part of any known systemic syndrome. Although chordoma occurrence in one family has been linked to chromosome 7q33,7 no gene mutation specific to chordoma has been identified. Chondrosarcomas are even more rare, comprising only 0.02% of all intracranial neoplasms.8 Although skull base chondrosarcomas can occur at any age, most patients present between 20 and 50 years of age9; the mean age at diagnosis is 40.7 years.10 Chondrosarcomas are slightly more common in men.11 Although they are usually isolated tumors, chondrosarcomas may occur in association with Paget disease, Ollier disease, or Maffucci syndrome.12,13 Chordomas occur at sites of embryonal remnants of the notochord: the sellar and parasellar region, clivus, spine, sacrum, and mediastinum. Thirty-five percent arise in the skull base, most commonly in the clival midline.1,8 Classification of the tumor location as superior, middle, and/or inferior clival is helpful in planning surgical approaches.14 With growth, chordomas expand and destroy bone and extend into extra-axial and intra-axial structures. Rostral notochord chordomas can often extend into the dorsum sellae and present in the sellar, suprasellar, or parasellar/ cavernous sinus areas. These may compress the pituitary gland, optic nerves and chiasm, and midbrain.15 Chordomas can extend ventrally through the clivus to present as a nasopharyngeal mass causing nasal obstruction or dysphagia.16 Chordomas extending from the dorsal clivus can compress the midbrain, pons, and medulla. Tumors extending dorsolaterally can involve the petrous temporal bone. Although chordomas are usually extradural, they may infiltrate and penetrate the cranial dura; dural invasion usually occurs late in the course of aggressive tumors. Surgical durotomies may facilitate the intradural extension of chordomas. Chordomas very rarely arise as primary intradural intracranial neoplasms.17,18 Metastasis usually occurs late in the course of the disease and is seen clinically in ~10 to 20% of cases.19 Metastatic potential has not been correlated with any histologic or clinical features. The most common metastatic sites are skin, bone, lung, and lymph nodes. Intradural metastases of skull base chordomas are rare, and most occur following surgery.20 Tumor recurrence may occur along routes of surgical access. Although metastases can be found in up to 40% of patients at autopsy,19 local tumor growth is usually the predominant threat to patient function and survival. Chondrosarcomas are mesenchymal, but their precise cell of origin is controversial; embryonal cartilaginous rests, mesenchymal pluripotent cells, and metaplastic fibroblasts have been proposed.10,21 Although they can occur anywhere in the skull base, most cranial chondrosarcomas occur at skull base synchondroses,22 notably those involving the clivus. Sixty-six percent are located at the petro-occipital junction (arising from the petroclival synchondrosis), 28% at the spheno-occipital synchondrosis, and 6% in the sphenoethmoid complex.12 Isolated chondrosarcomas are usually paramedian, but when part of Ollier or Maffucci syndrome, they may be midline.23 Chondrosarcomas originate as extradural tumors. Most grow slowly, destroy bone, and eventually extend into surrounding soft tissue. Most are low-grade malignancies12 and have a much better prognosis than do chordomas.24,25 The major concern after treatment is local recurrence.26 Metastases occur late, if at all, and affect no more than 10% of patients.11,22 Small chordomas and chondrosarcomas appear to arise within cranial bone, and with growth, they erode normal bone structures, displace the overlying periosteum and dura, and develop a nodular surface. They are not encapsulated, but the thinned sheath of expanded periosteum and dura often forms a pseudocapsule. Ultimately, these tumors erode cranial foramina and compress cranial nerves, brain, and basal arteries. Chordomas are grayish tan to bluish white, with a consistency that ranges from gelatinous to leathery and a texture that ranges from smooth to gritty. They may have soft foci of hemorrhagic necrosis or firm, calcium-dense areas. Histologically, chordomas are characterized by lobules and nests of large epithelium-like cells separated by fibrous strands. Tumor cells are arranged in sheets or cords in a background of myxoid stroma. They have abundant pink cytoplasm and moderately sized nuclei with mild to moderate atypia. Some cells have clear vacuoles, which impart a characteristic “bubbly” appearance to the cytoplasm. These “physaliferous” cells are large and vacuolated, and they contain mucus; they resemble the cells of the primitive notochord. Mitoses are limited and necrosis is common. Chordomas are immunohistochemically positive for cytokeratin, epithelial membrane antigen, carcinoembryonic antigen, and α-fetoprotein. Some chordomas stain positive for S-100 protein; this differentiates them from other sarcomatoid round cell or myxoid neoplasms.1 Staining for brachyury, SOX-9, and podoplanin (markers for primitive notochord) helps distinguish chordoma from chondrosarcoma.27 Chondrosarcomas are osteocartilaginous tumors with varying grades of malignancy. Macroscopically, they are nodular and gray to tan-white. The tumor consistency can be mucinous, firm, or gritty. Large yellow-white or chalky areas of calcification may be present. Chondrosarcomas erode cancellous bone and extend peripherally into soft tissue. Microscopically, four classic types of chondrosarcoma have been described: conventional, clear cell, dedifferentiated, and mesenchymal.12,28 Most skull base chondrosarcomas are of the conventional type. Conventional chondrosarcomas are composed of hyaline or myxoid cartilage or a combination of both. The neoplastic chondrocytes lie in clear lacunae within the hyaline matrix. Myxoid chondrosarcomas have bipolar or stellate neoplastic cells that float in a background of mucinous matrix. Tumor cells are arranged in a honeycomb of interconnecting strands and cords. They rarely have mitoses. As in chordomas, immunohistochemistry is positive for vimentin and S-100.12 In contrast to chordomas, however, chondrosarcomas typically do not express epithelial markers such as keratin and epithelial membrane antigen12 or markers for notochord differentiation.27,29,30 Tumors are graded histologically (1–3) based on cellularity, mitotic activity, and nuclear atypia. Tumor grade is predictive of invasiveness, growth rate, metastatic potential, response to therapy, and outcome.22,31–34 Grade 1 chondrosarcomas typically do not metastasize. Grade 2 chondrosarcomas have a higher metastatic tendency (10%), and grade 3 tumors have significant metastatic potential. The sellar and parasellar location of chordomas and chondrosarcomas presents formidable challenges to effective treatment. The relative inaccessibility of these areas and the propensity for local invasion of tumor with involvement of critical neural and vascular structures pose technical challenges to surgical resection. Despite such challenges, the combination of surgery and radiation can offer cure for most low-grade chondrosarcomas and provide significant control of chordomas and high-grade chondrosarcomas. Patients with a sellar or parasellar chordoma or chondrosarcoma typically present with headache and/or cranial neuropathies.1,10,12,35,36 Unilateral abducens nerve palsy is the most common clinical sign. Hearing loss, dysphagia, dysarthria, and tongue weakness occur when the tumor extends to the basiocciput and affects the middle and lower cranial nerves.9 Compression of the brainstem may manifest with sensory or motor signs. Endocrinopathies may result from involvement of the pituitary gland. Patients with chordomas have a poor prognosis if untreated, with a mean duration of survival ranging from 6 to 28 months.1,37 Patient age and gender, the histologic appearance of the tumor, the therapy received, and tumor recurrence have prognostic significance. Older patients generally do worse.36 In one series, all patients with tumors diagnosed at age 40 years or younger were alive 5 years later, in contrast to only 22% of patients with tumors diagnosed at age older than 40 years.38 Women have a poorer outcome than do men. A high mitotic rate also correlates with rapid growth and poor outcome. The natural history of chondrosarcomas is better, especially low-grade tumors. Nonetheless, most patients die without treatment.10 Histologic grade is strongly correlated with survival; patients with grades 1, 2, and 3 tumors had 5-year survival rates of 90%, 81%, and 43% respectively.33 The combination of surgery and radiation offers longer maintenance of neurologic function and survival. All patients warrant comprehensive evaluation, and most require aggressive treatment. Chordomas and chondrosarcomas are difficult to definitively distinguish radiologically.39 Chordomas are typically centered in the midline, whereas chondrosarcomas are usually paramedian. Both tumors may compress, displace, invade, or envelop critical surrounding structures, such as cranial nerves, cavernous sinuses, vessels of the circle of Willis, and the brainstem.40 Radiologic evaluation narrows the differential diagnosis and facilitates treatment planning (Table 9.1). Computed tomography (CT), in both the axial and coronal planes, determines the extent of osseous erosion and depicts intratumoral calcification. On soft-tissue-window CT scans, involvement of adjacent structures can be identified to some extent, although magnetic resonance imaging (MRI) is far superior in this regard. Chordomas often appear to be of relatively low density on CT owing to their mucoid content, and they typically enhance only minimally or mildly after the administration of iodinated contrast material. Chondrosarcomas are typically not as low in density as chordomas, and they are more likely to enhance moderately after contrast administration, often in a “honeycomb” or “popcorn-like” pattern. MRI is essential for accurate differential diagnosis and for defining the extent of the tumor (Fig. 9.1). Both chordomas and chondrosarcomas are usually hypointense or isointense to soft tissue on T1-weighted images and hyperintense on T2-weighted images.41 Chondrosarcomas are more likely to have a speckled pattern of signal voids, indicative of tumor calcification, although occasionally residual fragments of bone may appear as signal voids on MRI in chordomas. Enhancement following the administration of a gadolinium-based contrast agent is almost always present.42 Chordoma enhancement is variable and ranges from minimal to fairly intense; it often increases over time, such that the tumor will appear more intensely enhancing on delayed images. Chondrosarcoma enhancement is generally more intense than that of chordomas, and it can be heterogeneous owing to intermixed calcification. Tumor extension into the basal cisterns, cavernous sinuses, and adjacent regions such as the sella and nasopharynx is well delineated on MRI. The relationship of the tumor to the internal carotid and basilar arteries, especially any displacement or encasement, can be assessed by examining flow voids on MRI.43 Angiography10 with test balloon occlusion may be indicated if vessel sacrifice is contemplated. Various options exist for the management of sellar and parasellar chordomas and chondrosarcomas. These include clinical and radiologic observation, biopsy followed by observation, biopsy followed by radiation, surgical removal, surgical removal followed by radiation, and chemotherapy. The appropriate choice of treatment requires confidence in the diagnosis, familiarity with the various treatment options, and predicted treatment outcome in the context of the individual patient. The diagnosis of the tumor is usually based on characteristic radiographic features, as discussed above, although distinguishing chordoma from chondrosarcoma may not be possible without tissue sampling. Occasionally, a preliminary biopsy is indicated if the patient’s age, medical condition, and neurologic deficits or the tumor’s size and location contraindicate surgery and only radiation therapy will be offered. Additionally, a biopsy should be performed if other possibilities, such as metastasis, pituitary adenoma, and lymphoma, cannot be excluded on the basis of imaging alone. In this situation, the biopsy can often be performed at the start of an intended resection. Because chordomas and chondrosarcomas are predominantly extradural, standard stereotactic needle biopsy techniques may not be useful, and biopsy is more likely to be successfully performed through the nose. Extensive resection and high-dose radiotherapy for both chordomas and chondrosarcomas are associated with better tumor control and longer patient survival10,24,25,36,44 than is observation, surgery alone, or radiation alone. Patients should be evaluated by a multidisciplinary treatment team that includes a neurosurgeon, an otolaryngologist, and a radiation oncologist. A comprehensive treatment plan that maximizes tumor control and minimizes iatrogenic complications should be fashioned for each patient. Surgery is generally indicated as the first step toward the restoration or preservation of neurologic function, with radiation therapy to follow. The surgical approach should be tailored to the goal chosen for each individual patient. The goal of surgical resection may vary from complete en bloc excision to piecemeal gross total resection of the tumor to radical subtotal removal that decompresses critical neurovascular structures and improves tumor geometry for postoperative radiotherapy. The choice of the surgical approach depends on the size, site of origin, and direction of expansion of the tumor; involvement of critical neurovascular structures; prior treatment; the patient’s overall health; and the surgeon’s technical expertise with the approach. Surgery is usually the initial therapeutic modality for chordoma and chondrosarcoma. It is indicated to obtain diagnostic tissue, relieve neurologic symptoms, and resect tumor. Advances in skull base surgery have improved surgical outcome with respect to extent of resection, neurologic morbidity, and cosmesis (Fig. 9.2). For some tumors, staged operations via multiple approaches may be required to obtain satisfactory tumor exposure and removal. Microscopic and endoscopic techniques may both be helpful.45 If reoperation is being planned, the sequelae of the earlier operation, such as scar tissue and distortion of the normal anatomy, must be taken into account. A fresh route of access is often preferred. Many chordomas and chondrosarcomas of the sella and parasellar region pose significant challenges to complete resection by virtue of extensive bone involvement and their proximity to or envelopment of critical neural and vascular structures. The basic strategy is to maximize exposure of tumor and involved critical structures with minimal brain retraction. Coordination between neurosurgeons and otolaryngologists can achieve both improved exposure by traversing air sinuses or removing bone and more secure repair of the skull base defect with vascularized soft tissue flaps, which minimize the risks for cerebrospinal fluid (CSF) leakage and infection. The surgical approaches to chordomas and chondrosarcomas of the sella and parasellar region can be grouped according to whether they are exclusively midline or also have lateral extension46 (Table 9.2). An extradural approach is generally used because most of these tumors are extradural.47 This allows access to the bone of origin. When the overlying dura is infiltrated, the involved bone must be removed to approach the infiltrated dura. Tumor-infiltrated bone should always be drilled away, even if it is not restricting access to dural and intradural tumor. Tumors limited to the sella and the immediately adjacent suprasellar, infrasellar, and cavernous sinus areas can be approached by a simple transsphenoidal approach.48,49 A transnasal approach, or a combination of transnasal and transoral approaches, can be used to access the inferior midline extension of tumor down to the C1 vertebra. Suprasellar midline tumor extension that lies between the supraclinoidal internal carotid arteries can be accessed if the transsphenoidal approach is extended by removing the tuberculum sellae and adjacent planum (transtubercular, transplanum). Even large, posteriorly directed intracranial extensions of the tumor that grow posteriorly through the dura to displace the brainstem can be removed by such extended transsphenoidal approaches.%50–56 Rigid endoscopes or the operating microscopes can be used. The endoscope’s wide-angled view, illumination, and high magnification improve visualization of the surgical anatomy, and angled endoscopes can extend the surgical field laterally, superiorly, and inferiorly. Because this endoscopic view is two-dimensional, it requires supplementation with tactile and proprioceptive clues to depth (Table 9.2). Suprasellar extension of firm tumor that extends lateral to the internal carotid arteries or incorporates cranial nerves or intracranial vessels usually warrants a craniofacial procedure. This combines the suprasellar exposure of a frontal craniotomy with the clival access of a transnasal, transsphenoethmoid approach. This approach can be endoscopically assisted. The transbasal and extended subfrontal approaches attempt to achieve a similar exposure from a single perspective.14,57 Parasellar tumors extending inferiorly in the midline and paramedian lower clivus are accessible through a transnasal, transsphenoidal, transclival approach. Even further inferior extension to the craniovertebral junction, C1, and dens requires the addition of a transodontoid approach, which is usually transoral.58 Tumors with significant lateral and inferior extension require the addition of a more oblique approach, such as the posterolateral or far lateral approach.59 The lateral extension of parasellar and midclival tumors anterior to the internal carotid arteries may be accessed by adding various degrees of maxillotomy to a more midline transnasal (endoscopic) or transfacial (microscopic or endoscopic) approach.60 More posterior tumors with parasellar extension, especially when superior or lateral to the cavernous internal carotid artery, usually require a frontotemporal craniotomy; this may include dissection of the involved cavernous sinus.61,62 Parasellar tumor involving the petrous apex, tentorium, and subjacent posterior fossa can be exposed by a subtemporal, anterior transpetrosal route.63,64 Larger tumors extending medially, inferiorly, and posteriorly usually require one of the posterior transpetrosal approaches. A presigmoid exposure is common to this group, but the extent of labyrinthectomy is dictated by the location, size, and consistency of the tumor and the status of the patient’s hearing.65 These lateral approaches have the advantage of avoiding the potentially contaminated nasal sinuses and oropharynx. In some cases, the risks for CSF leak and infection can be reduced by using a two-stage procedure: a lateral approach for removal of the intradural lateral tumor and dural repair followed by an anterior approach for the sellar midline tumor. Chordomas often have two intermixed components: a soft, gelatinous portion within expanded bone or dura and a more sinewy part infiltrating and expanding the dura or extracranial soft tissue. The gelatinous part can be easily removed by suction or gentle dissection and curettage. The surrounding thickened arachnoid usually protects cranial nerves, brainstem, and vessels, except in reoperations, when this protection has been previously transgressed. In these reoperations, the tumor may surround cranial nerves and extend between brainstem arteries and the pia. Aggressive resection then risks cranial nerve injury and brainstem stroke from injury to perforating arteries. The sinewy portion requires more sharp dissection because the plane between tumor and the surrounding soft tissue is often obscured by tumor invasion. Whenever possible, thickened and thus potentially infiltrated dura and extradural soft tissue should be excised. Chondrosarcomas are more discrete and thus often are more completely resectable than chordomas. Some chondrosarcomas may be heavily calcified and firmly fixed to the skull. These can be removed only by fragmenting them or by drilling. Before these calcified fragments are manipulated, it is essential to first identify and dissect tumor from adjacent cranial nerves and critical vessels. Incorporation of cranial nerves or vessels within the calcified tumor may warrant subtotal resection to preserve function. Adherence to some basic principles helps reduce the incidence of surgical complications (Figs. 9.3, 9.4, and 9.5). First, removal of bone is almost always preferable to retracting brain as a means of gaining exposure, with the exposure only as large as is required to remove the tumor. The more extensive the dissection, the greater is the risk to cranial nerves, vessels, and dura (Fig. 9.3). Second, for tumors with intradural extension, the dural defect must be closed securely, especially when the approach traverses the nose or oral cavity. Removal of involved bone and dura in transnasal, expanded, extended transsphenoidal approaches can often be complicated by CSF rhinorrhea and meningitis.56 Historically, these occurred in 10 to 50% of patients who had tumors with a significant intradural component. These complications can be minimized by a meticulous, multilayered closure, preferably one in which at least one layer is vascularized. Dural openings should be sutured primarily if possible, or closed with autologous fascia or dural substitute if primary closure is not feasible. If a graft cannot be sewn in, an inlay graft of dural substitute or fascia should be placed such that its edges lie deep to the margins of the dural opening. This is then covered with a thin layer of tissue adhesive followed by an onlay graft of fascia or dural substitute. This onlay graft can be held snugly in gasket seal fashion by a firm plate (thin bone or cartilage, polyethylene glycol plate, or titanium wire mesh) wedged beneath the margins of the bone opening66 (Fig. 9.4). The plate can then be covered with more tissue adhesive followed by vascularized soft tissue, such as the Hadad-Bassagasteguy flap (nasal septal pedicled mucosa fed by the posterior nasal artery)67 in transnasal approaches, pericranium for subfrontal craniotomies, or temporalis fascia and muscle for lateral approaches (Fig. 9.4). Insecurely repaired large openings mandate temporary drainage of CSF through a ventriculostomy or lumbar drain. Third, dissection should be meticulous. Approaches to these tumors frequently require maneuvering around the internal carotid and basilar arteries and cranial nerves. Adherence to microsurgical techniques, facilitated by endoscopic illumination, magnification, and angulation, permits safe tumor removal (Fig. 9.5). Inadvertent injury to the basilar and internal carotid arteries or their branches can be catastrophic. Similarly, the greatest care must be taken to avoid injury to the cranial nerves. Close attention to anatomic keys is essential.60 Neuronavigation techniques are very helpful.68 The pseudoencapsulated nature of both chordomas and chondrosarcomas and the soft consistency of the intradural extensions of chordomas facilitate separation of tumor from nerves and blood vessels. These structures are more likely to be displaced than invaded. The exception occurs at neural foramina, where nerves, surrounded by tumor-infiltrated bone, are most vulnerable to injury. Here and in the cavernous sinuses, it is sometimes preferable to leave small deposits of tumor rather than risk injury to a cranial nerve. Intentional sacrifice of a cranial nerve is almost never justified by a desire to obtain a complete resection of tumor because residual tumor can be treated by postoperative radiation therapy. Fig. 9.3 Removal of a clival chordoma. (A) Extradural tumor (depicted radiographically in Fig. 9.2) is removed, and the mid clivus is drilled away. (B) The dura is opened, and intradural tumor is debulked. (C) Tumor is dissected from the brainstem. (D) Pons, after tumor removal, with basilar artery traversing inferiorly. Fig. 9.4 Repair after resection of a clival chordoma. (A) Tumor specimen (removed as shown in Fig. 9.3). (B) Inlay graft of dural repair matrix. (C) A gasket seal closure with dural repair matrix and a polyethylene glycol plate. (D) A pedicled septal mucosal flap placed over the closure. Fourth, the intent of surgery, as formulated preoperatively, must always be kept in mind. Not infrequently, the goal of surgery is to remove sufficient tumor to optimize the geometry of the tumor bed for subsequent radiation therapy. Because radiation exposure of the brainstem or optic pathways is the most common factor limiting the dose that can be given to these tumors, the primary indication of surgery is the removal of tumor invaginating the brainstem and in proximity to the optic nerves or chiasm (Fig. 9.6). Reduction of tumor mass is helpful in reducing radiation target volume, but the advantage conferred by reducing the tumor burden decreases at low tumor volumes69 (Table 9.3). This diminishing return, coupled with the relatively indolent growth of low-grade chondrosarcomas and the microscopically invasive nature of chordomas and of higher-grade chondrosarcomas, makes aggressive efforts at total resection ill advised (Fig. 9.7). Most studies of skull base chordomas and chondrosarcomas offer evidence that surgical resection helps extend survival.22,36,57,70–76 Jawad and Scully70 reviewed 962 treated chordomas. They found that size less than 8 cm, age younger than 59 years, Hispanic ethnicity, and surgical resection were all independent predictors of longer survival. Durations of survival were very similar for patients with “inoperable” disease and those for whom “surgery was recommended but not performed,” suggesting that the benefits of surgery were not necessarily attributable to selection bias. In another series of 51 patients with intracranial chordomas, the 40 patients whose tumors were resected (partially or completely) were more likely to survive 5 years (55%) than were the 11 whose tumors were only biopsied (36%).36 A more complete resection may also be associated with a better outcome than a less aggressive approach.10,74 There are a few reports of long-term disease-free survival following radical resection alone.10,77 These studies support attempting radical surgical resection in all patients in whom it is feasible. Some surgical series in which radical resection was followed by radiation therapy report even better rates of tumor control. In one series of 60 patients (46 chordomas and 14 low-grade chondrosarcomas), no tumor recurrence was found in 80% at 3 years and 76% at 5 years after surgery.10 Patients with chondrosarcomas did better than those with chordomas (5-year recurrence-free survival rates of 90% and 65%, respectively). However, 11 patients died during the postoperative follow-up period: 3 of systemic complications within 3 months of surgery, 5 of tumor recurrence, 1 of unrelated causes, and 2 of late complications of radiotherapy. The rates of nonlethal complications, such as CSF fistula, meningitis, and cranial nerve deficits, were also significant. This experience suggests that aggressive attempts to remove all tumor must be justified by both longer patient survival and avoidance of serious neurologic and functional compromise.10,44 Morbidity and mortality rates for surgical resection of chondrosarcomas are similar to those for chordomas. Some authors argue that complete excision of a low-grade chondrosarcoma obviates the need for postoperative irradiation. Among patients who underwent complete resection of their tumor but not postoperative radiation therapy, 78.3% experienced 5 years of recurrence-free survival.25,78 A superior outcome for even completely resected tumors following surgery and high-dose radiation therapy argues for the use of adjuvant radiotherapy. Fig. 9.6 Postoperative CyberKnife plan for chordoma. The resection field in the same patient shown in Figs. 9.2 through 9.4 was treated with 35 Gy in five fractions, the equivalent of 80 Gy in 2-Gy fractions. Dose to the brainstem surface was less than 22 Gy in five fractions, the equivalent of 35 Gy in 2-Gy fractions. The risk for microscopic residual tumor even after complete gross tumor resection is very high for almost all chordomas and for many chondrosarcomas. Local regrowth of these tumors in the skull base is not unusual, even after aggressive surgical treatment. Postoperative radiation therapy targeting the entire tumor volume is therefore indicated for all chordomas, incompletely resected low-grade chondrosarcomas, and higher-grade chondrosarcomas. Multiple clinical series document longer survival of patients with this approach.1,5,6,10,36,79–87 One meta-analysis of 464 chordomas with a mean follow-up of 39 months found that younger age, chondroid histology, and treatment with surgery and radiation correlated with lower recurrence rates.79 The efficacy of radiation therapy of chordomas and chondrosarcomas is highly dependent on the dose delivered.35,88,89 Lower doses of 45 to 60 Gy are associated with poor rates of progression-free and overall survival. Recurrence rates of 50 to 100% have been reported in chordomas treated with conventional irradiation.85,86 Higher-dose radiation (66–84 Gy) can delay or prevent recurrence. The goal is to deliver these higher doses in a highly conformal way to maximally target tumor volume but spare adjacent radiosensitive structures from exposure beyond their tolerance. Exclusion of the optic nerve and chiasm at 55 to 60 CGE90 (cobalt gray equivalent) may protect vision, and exclusion of the pituitary at 50 CGE may prevent endocrinopathies.91 Irradiation techniques include intensity modulation radiotherapy, conventionally fractionated radiotherapy, external beam photon radiation, particle beam radiotherapy, and stereotactic radiosurgery. Intensity modulation radiotherapy modulates the dose intensity pattern of conventionally fractionated radiation to match the tumor shape. In one series of chondrosarcomas, postoperative fractionated radiotherapy provided a 5-year recurrence-free survival rate of 100%.92 Takahashi et al81 reported the results of carbon ion radiation therapy in 9 of 32 patients following aggressive resection of a skull base chordoma. The 3-year recurrence-free survival rate following carbon ion therapy was 70%, compared with 57.1% after conventional radiotherapy and 7.1% in the untreated group. Conformal proton radiation has been delivered to chordomas in high doses ranging from 66.6 to 79.2 CGE (median, 68.9 CGE).87,92,93 A recent review of 416 chordomas treated with proton beam therapy in seven uncontrolled single-arm studies87 found better outcomes and fewer complications for proton beam than had been reported for conventional radiation. In the largest single-institution proton beam series, the 5-year local tumor control rate was 54% for 125 chordomas and 98% for 130 low-grade chondrosarcomas.93 Favorable prognostic factors for the long-term control of chordomas were male gender, small tumor volume, and high minimum dose. In another study, mixed photon-proton beam irradiation of chordomas to 75.6 to 82.9 CGE yielded 5- and 10-year local recurrence-free survival rates of 64% and 42%, respectively.94 For chondrosarcomas treated with 66 to 83 CGE, the 5- and 10-year local recurrence-free survival rates were 97% and 92%, respectively.94 In these proton series, relapse was overwhelmingly local. After local relapse, the actuarial rates of survival for patients with chordomas were 44% at 3 years and 5% at 5 years.95 Stereotactic radiosurgery targets high doses of X-rays from a linear accelerator (LINAC) or CyberKnife or gamma rays from cobalt sources (gamma knife) to a tumor in one to five treatment sessions. This precisely targets tumor very close (within 2–3 mm) to critical structures96 (Fig. 9.8). Because the risk for injury rises with targeted volume, radiosurgery is usually limited to tumor volumes less than 10 cm3. Radiosurgery may be particularly useful in treating small unresectable residual or recurrent chondrosarcomas or chordomas.97 Henderson et al82 reported excellent outcomes following CyberKnife therapy of 24 tumors. They recommended a dose of 40 Gy in five sessions to the clinical treatment volume, defined as the gross tumor volume and a 1-cm margin. In another radiosurgical series including both types of tumor, 3 of 15 patients died of local tumor recurrence outside the target volume, and one died of intercurrent disease. Of the 11 surviving patients, 10 had tumors that were unchanged or smaller during the mean 4-year follow-up period. No neurologic or endocrine toxicity was observed.96 This series highlights the challenges these tumors pose for radiosurgery. Unlike benign encapsulated tumors (eg, meningiomas and vestibular schwannomas) or metastases (which are usually spherical), chordomas and chondrosarcomas are irregularly invasive. These tumors are often quite large at diagnosis and even after aggressive resection may present a large volume at risk for recurrence. The microscopic and even gross tumor limits can be challenging to define radiographically. Once the target is determined, the irregular shape of the tumor and limits created by adjacent critical structures (optic pathways, brainstem) require complicated multiple isocenter treatment plans. Longer follow-up of larger number of patients is needed to confirm the role of radiosurgery in these tumors with a marked propensity to recur locally. The risk for neurologic damage, such as brainstem injury and death, visual field loss, and hypopituitarism, is associated with all modalities of radiation therapy.69,91,98 Although newer, more conformal radiation strategies improve efficacy and lower morbidity, local recurrence is common, and many patients, especially those with chordomas, will suffer from the side effects of treatment. Almost all chordomas and some chondrosarcomas recur despite radical resection and radiation therapy. In most cases, recurrence will eventually prove fatal. When tumors recur, radiotherapy is offered for those that have not been previously irradiated. Repeat resection to reduce the tumor volume and alleviate brainstem compression may be needed. Surgery for previously resected tumors carries a higher incidence of complications,10 but when previously irradiated tumors recur, repeat surgical resection is often the only choice. In one series, patients with relapse of any kind after proton radiation had actuarial survival rates of 43% at 3 years and 7% at 5 years. Local relapse carried actuarial survival rates of 44% at 3 years and 5% at 5 years and distant recurrence rates of 25% and 12%, respectively. Reoperation on 49 of 60 patients with locally recurrent tumor produced disease stabilization in 26 (53%) and yielded actuarial survival rates of 63% at 2 years and 6% at 5 years, compared with a 2-year survival rate of 21% for those receiving only supportive care.95 Therefore, re-operation is of value in surgically accessible tumor recurrence, even though it is associated with greater risks and has less survival benefit than primary surgery. Stereotactic radiosurgery, too, may be useful for tumor recurrence. Ito et al80 recently reported using gamma knife therapy in patients who had recurrent disease following aggressive resection of a clival chordoma. Among 19 patients who underwent surgery, 11 had recurrence. Patients with recurrent tumors, some of whom received reoperation, underwent gamma knife treatment. At a mean follow-up of 71 months, tumor control was achieved in all 11 patients with recurrence. Chemotherapy is usually reserved for surgically inaccessible, previously irradiated recurrent tumor or for metastatic disease. As advances in surgery and radiotherapy have improved rates of local control, the effective treatment of metastatic disease has become increasingly important. Up to one-third of patients with chordomas develop distant metastases,19,99,100 most commonly to the lungs, liver, and bones.2 Historically, chemotherapy has had poor efficacy.101 Newer agents such as imatinib mesylate, a tyrosine kinase inhibitor that targets several enzymes and a growth factor expressed in chordomas, may offer improved outcomes.102 A recent case report noted marked regression of a recurrent chordoma following intratumoral injection of carboplatinum.103 Chordomas and chondrosarcomas of the sellar and parasellar region are rare (Table 9.4). The relative inaccessibility of these areas of the skull base, the proximity of the tumors to critical neurovascular structures, their relative insensitivity to radiation and chemotherapy, and their propensity to recur locally make them very challenging to treat. Treatment strategies are complex and require meticulous planning and execution of surgery and high-dose radiation therapy. Such an approach can offer cure for almost all low-grade chondrosarcomas and provide meaningful control of chordomas and high-grade chondrosarcomas.
 Incidence and Epidemiology
Incidence and Epidemiology
 Location
Location
 Pathology
Pathology
 Challenges
Challenges
 Clinical Presentation
Clinical Presentation
 Natural History
Natural History
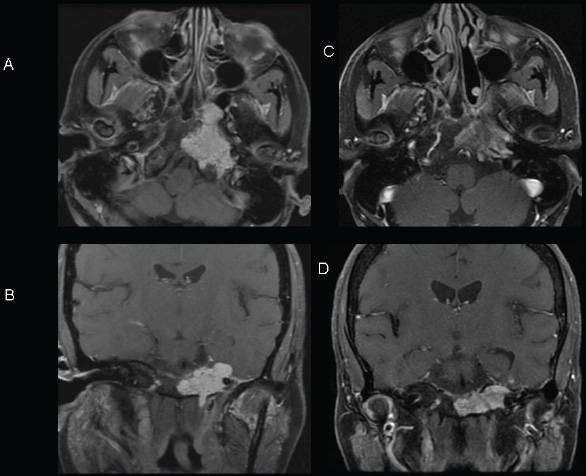
 Radiologic Investigations
Radiologic Investigations
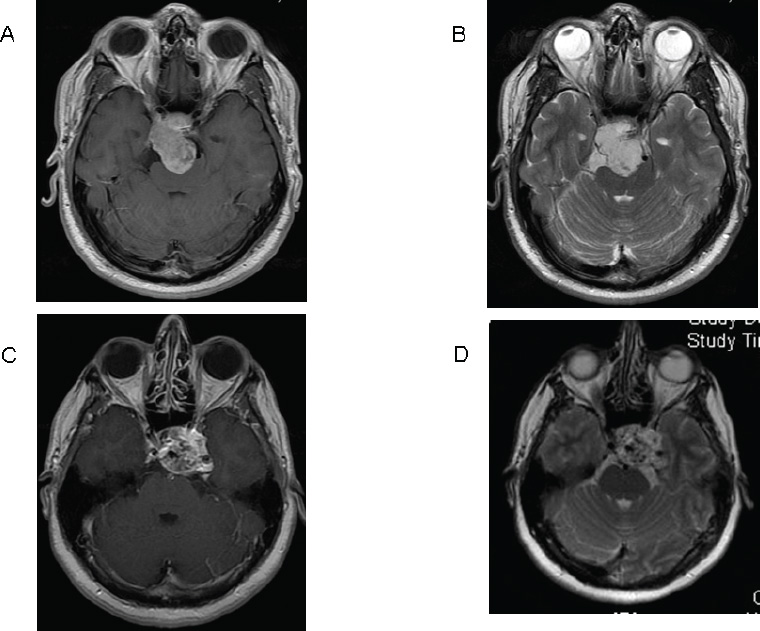
 Treatment
Treatment
 Surgery
Surgery
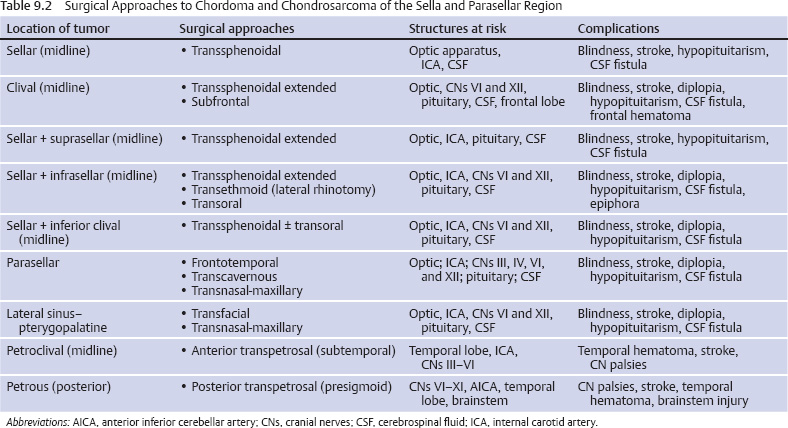
Surgical Approaches
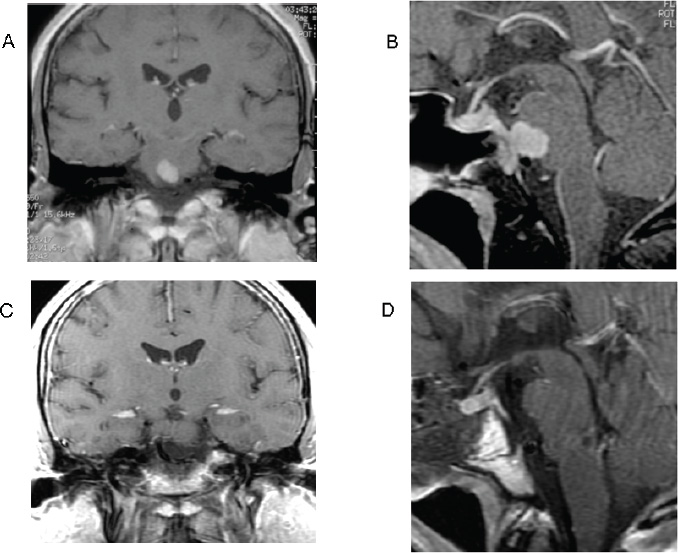
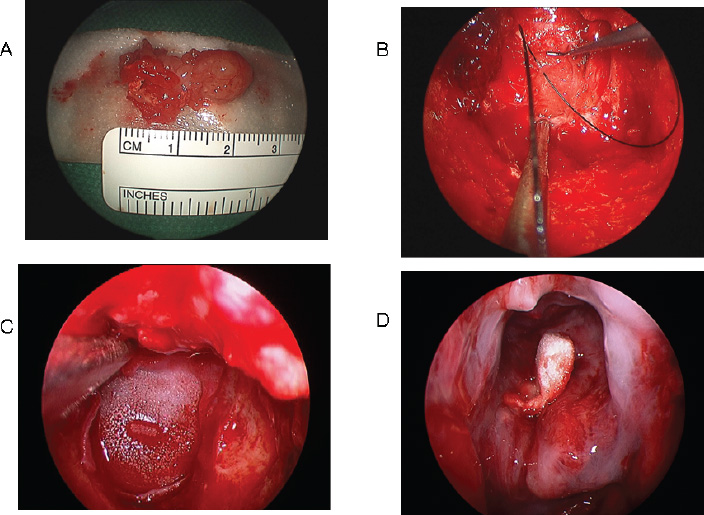
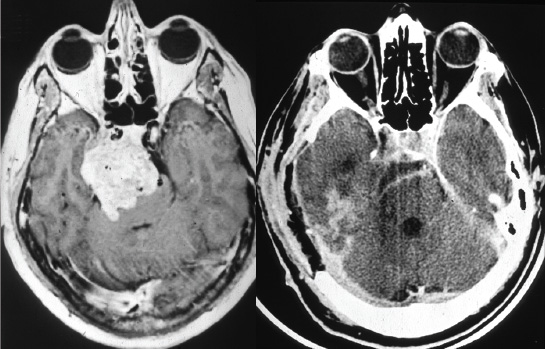
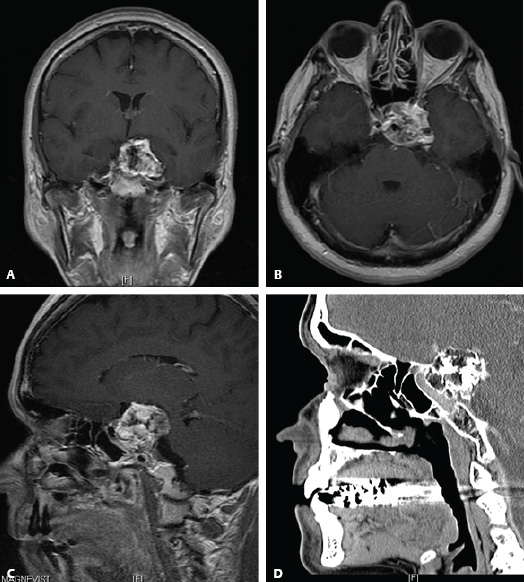
Surgical Technique
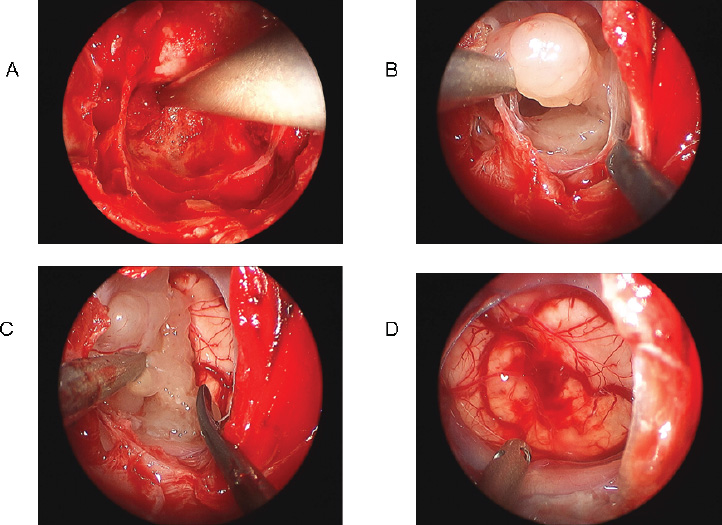
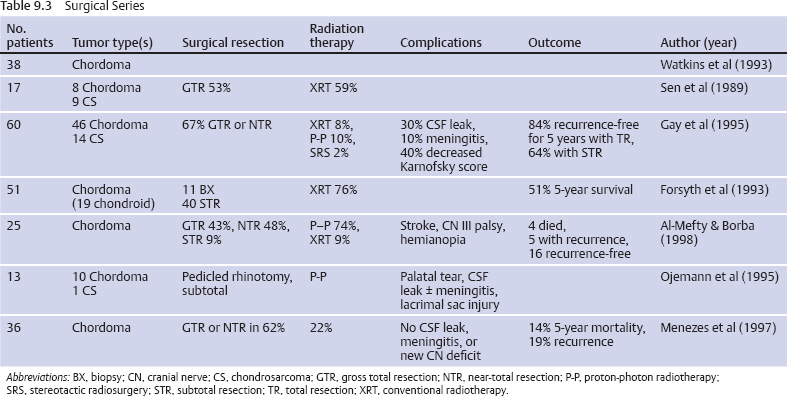
Surgical Outcomes
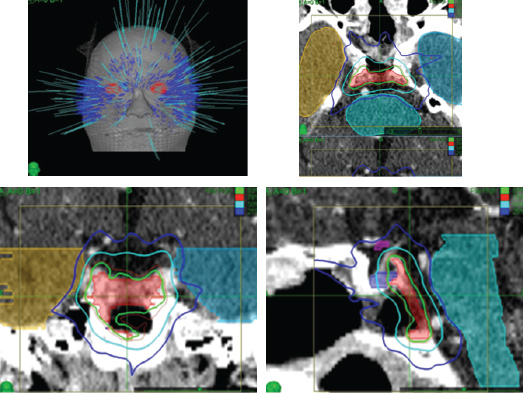
 Radiation Therapy
Radiation Therapy
 Tumor Recurrence
Tumor Recurrence
 Chemotherapy
Chemotherapy
 Conclusion
Conclusion

References
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


