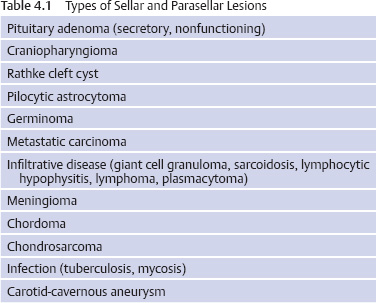
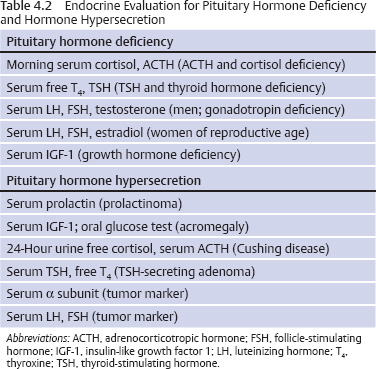
4 Medical Evaluation and Management Of the many types of lesions that occur in the sella or parasellar region, a pituitary adenoma is the most common. All lesions in this area require an endocrine evaluation to determine if the lesion is a secretory pituitary adenoma and if so what type of hormone is oversecreted. Also, even if the lesion is not a pituitary adenoma, a comprehensive endocrine evaluation will assess the pituitary function and whether any hormonal replacement is required. An ophthalmologic evaluation is indicated in patients with a lesion approximating or compressing the optic apparatus (optic nerves, optic chiasm). The neuro-ophthalmologic evaluation is covered in Chapter 5. The endocrine evaluation is accomplished with measurement of the serum hormone levels and, for the situation of possible Cushing disease, a 24-hour urine test for urine free cortisol. Additional dynamic testing may be required in patients with possible Cushing disease or acromegaly—all of which can be accomplished in the outpatient setting. The important concept is that the pituitary and the adjacent area comprise both an anatomic and a functional entity, so that a combined evaluation of the anatomy and physiology is required to determine the appropriate treatment or treatments. Table 4.1 details the most common types of sellar and parasellar lesions that should be considered when each patient is reviewed. A lesion in the sella often causes loss of pituitary function and/or exertion of a mass effect. Depending on the location, mass effect can cause loss of vision (eg, visual field defect, decreased visual acuity), cranial nerve dysfunction (eg, diplopia, ptosis), or headache. A lesion that causes acute visual loss requires immediate evaluation and treatment. Endocrine consequences of a pituitary lesion affect the reproductive system most commonly.1 Thus, men often have symptoms of hypogonadism—decreased libido, erectile dysfunction, and infertility. Women of reproductive age may develop irregular menses, amenorrhea, or infertility. Adolescents may experience delayed or arrested puberty. Another common pituitary deficiency is loss of growth hormone (GH) secretion, resulting in fatigue in adults and arrested or decreased growth velocity in children and adolescents. The two most important hormones are cortisol and thyroid hormone, both of which are necessary for life. Symptoms of cortisol deficiency include fatigue, headache, weight loss, diminished appetite, and in some patients hypotension and syncope. Symptoms of thyroid hormone deficiency include fatigue, cold intolerance, weight gain, constipation, difficulty concentrating, and memory problems. Deficiency of the posterior pituitary hormone vasopressin causes polyuria (particularly nocturia), polydipsia, and potentially volume depletion if the patient does not drink an adequate amount of fluids. This condition is termed diabetes insipidus (DI), which is rare in patients with a pituitary adenoma; if there are symptoms, the possible diagnoses are a craniophyaryngioma, lymphocytic hypophysitis, sarcoidosis, or metastatic carcinoma. Excessive hormone secretion causes symptoms and signs associated with overproduction of prolactin, growth hormone (acromegaly), or adrenocorticotropic hormone (ACTH; Cushing disease). A prolactin-producing adenoma is the most common type of secretory pituitary adenoma. Clinical features in women of reproductive age with a prolactinoma include irregular menses, amenorrhea, or infertility; galactorrhea may or may not be present. Men with a prolactinoma develop diminished libido, erectile dysfunction, and infertility; gynecomastia and galactorrhea may also occur. Because many men and postmenopausal women seek medical attention late in the course of disease, visual loss may be the presenting feature. Features of Cushing disease, which is excessive cortisol production stimulated by an ACTH-producing pituitary adenoma, include weight gain, diabetes mellitus, hypertension, osteoporosis, bone fractures, depression, and memory loss. The least common type of secretory pituitary adenoma is the thyroid-stimulating hormone (TSH)–secreting tumor, which causes clinical or subclinical hyperthyroidism (weight loss, tachycardia, frequent bowel movements, and anxiety). Table 4.2 details the necessary hormone tests to evaluate a pituitary or parasellar lesion. The most important issue is to determine if the patient has secondary adrenal insufficiency and/or secondary hypothyroidism because these hormones are necessary for essential life functions. Low morning serum cortisol and ACTH levels may be adequate to diagnose secondary adrenal insufficiency. If there is doubt, a stimulation test such as an ACTH stimulation test or insulin-induced hypoglycemia is indicated. The insulin hypoglycemia test is the most rigorous and reliable study; this test is also the most accurate for diagnosing GH deficiency. However, the insulin hypoglycemia test must be monitored by a physician and is contraindicated in patients with coronary artery disease, a seizure disorder, or general debility. For thyroid hormone deficiency, both the serum free thyroxine (free T4) and TSH levels should be measured. A low serum free T4 is the most reliable test. Patients with pituitary disease may have a “normal” serum TSH in the setting of a low free T4 level. DI is primarily a clinical diagnosis (polyuria, polydipsia, excessive thirst, and in particular frequent nocturia—ie, urination every 30–60 minutes during the night). Serum sodium and serum osmolality are normal if the patient has intact thirst sensation and no restriction of fluid intake; serum osmolality may be normal, but the urine specific gravity should be low. Gonadotropin deficiency in men is diagnosed by clinical symptoms and by measuring the serum testosterone and luteinizing hormone (LH). In gonadotropin deficiency, the serum testosterone level will be low in the setting of a “normal” serum LH (not normal for low testosterone). In women of reproductive age, irregular menses, amenorrhea, and infertility are the best indicators of gonadal dysfunction. Serum estradiol may be low or “normal” depending on the stage of the menstrual cycle, and serum LH is usually in the “normal” range, but lack of regular menses or ovulation indicates gonadal dysfunction. GH deficiency is diagnosed by either a low serum insulin-like growth factor 1 (IGF-1) level in the setting of several pituitary hormone deficiencies or by a subnormal GH response to a stimulation test (eg, insulin-induced hypoglycemia or arginine infusion). Serum prolactin should be measured to determine if a pituitary lesion is a prolactinoma, because medical therapy with a dopamine agonist is the first line of treatment for a prolactinoma. The prolactin level must be correlated with the size of the pituitary or parasellar lesion. Any lesion in this region may cause a mild elevation of serum prolactin (interference with the prolactin inhibitor hormone, dopamine, through the pituitary stalk). As a general rule, in the setting of a macroadenoma (>10 mm), the serum prolactin level should be greater than 200 ng/mL for a true prolactinoma. This assessment is necessary to determine the course of treatment—medical therapy or surgery. One caution: In the setting of an elevated serum prolactin level (>200ng/mL), the serum specimen must be diluted to obtain an accurate value (prolactin >200 ng/mL is not sufficient), and the physician must request that the laboratory perform the necessary dilutions to obtain the actual value. A value of >200 ng/mL may be 2000 or 20,000 or greater. To assess the response to medical therapy, an accurate baseline prolactin value is also required. Acromegaly is diagnosed by clinical features, an elevated serum IGF-1 level, and a serum GH level that does not decline to >1 ng/mL after oral glucose (75 or 100 g). There are important considerations about these tests. Although the IGF-1 assay is reliable and reproducible, over the past 2 years there have been false elevations of IGF-1 related to the database for the range of normal according to age. This problem is being addressed but has not yet been fully resolved. The definitive test for acromegaly is the GH response to an oral glucose challenge (oral glucose tolerance test, or OGTT). The test must be performed correctly to interpret the results. Baseline serum glucose and GH are measured, the patient drinks a glucose solution (75 or 100 g), and the serum glucose and GH levels are measured every 30 minutes for 2 hours. The current guideline for a normal response is a serum GH level of >1 ng/mL. Some patients with acromegaly may have a paradoxical increase in GH.2 Cushing disease is diagnosed by demonstrating a consistent overproduction of cortisol in the setting of detectable or elevated serum ACTH. Cushing disease is the most problematic of all pituitary adenoma diagnoses for several reasons, including overlap of the clinical features with those of other disorders (polycystic ovarian syndrome, obesity, depression) and the variable sensitivity and specificity of tests. Consistent overproduction of cortisol is demonstrated by three types of screening tests: elevated 24-hour urine free cortisol (preferably measured by tandem mass spectrometry), loss of circadian rhythm with elevated nighttime salivary cortisol levels, and failure of the serum cortisol to decline to >1.8 μg/dL at 8 AM after ingestion of dexamethasone at 11 PM the previous night.3 These three types of screening tests are ~92% accurate; thus, repeated tests may be necessary to establish the diagnosis. Serum ACTH may be in the normal range or elevated. The traditional high-dose dexamethasone suppression test (8 mg overnight or 2 mg every 6 hours for 48 hours) was developed to distinguish between pituitary-dependent or ectopic ACTH syndrome. Unfortunately these tests are not sufficiently sensitive or specific to exclude ectopic ACTH production by a tumor in the lung, pancreas, or thyroid gland. Because ~50% of patients with a pituitary adenoma causing Cushing disease have no visible lesion on magnetic resonance imaging (MRI) with a pituitary protocol and because 10% of adults with normal pituitary function have a visible lesion in the pituitary gland (“incidentaloma”), an MRI study is not sufficient to recommend pituitary surgery. The inferior petrosal sinus sampling (IPSS) study is the most precise method to determine if the source of ACTH is the pituitary gland and to exclude ectopic ACTH syndrome. This test involves comparing the central (petrosal sinus, left and right) and peripheral (inferior vena cava) ACTH levels before and after the administration of corticotropin-releasing hormone (CRH). A ratio of the basal central to the peripheral ACTH level of >2 or a CRH-stimulated ratio of >3 indicates a pituitary etiology. This invasive study should be performed only by an experienced interventional radiologist or neuroradiologist; cannulation of the inferior petrosal sinuses requires appropriate experience and expertise. This study is not without risk, including thrombosis and stroke, emphasizing the requirement that it be performed by an experienced radiologist. An uncommon type of secretory adenoma produces excessive α subunit, which causes no specific clinical features but often causes hypogonadism. Measurement of serum α subunit serves as a tumor marker before and after surgical removal of the adenoma. Although many pituitary adenomas are gonadotrope tumors by immunohistochemical criteria, they rarely secrete excessive amounts of LH or follicle-stimulating hormone (FSH). Measurement of the serum LH and FSH serves as a tumor marker in the event the tumor produces an excessive amount of one or both hormones. A diagnosis of hormone deficiency and/or hormone hypersecretion directs treatments. As noted, a prolactin-producing adenoma is treated medically with a dopamine agonist drug. Other types of lesions are most commonly treated by resection, radiosurgery, radiation therapy, and even chemotherapy. Replacement of cortisol and/or thyroid hormone is necessary before a therapeutic intervention. After surgery or another treatment, patients should be re-evaluated for pituitary hormone deficiency. In the case of patients with acromegaly or Cushing disease, serum GH and/or cortisol immediately after surgery is a helpful indicator of the outcome of the operation. Periodic and comprehensive re-evaluations of the endocrine status of patients should be performed to detect for tumor recurrence in the case of secretory pituitary adenomas. In addition, extended endocrine follow-up should be performed to detect hypopituitarism, which can arise months to years after intervention.4 Delayed hypopituitarism has been observed following radiosurgery and radiation therapy for sellar and parasellar disease.5 All lesions in the sellar or parasellar region require an endocrine evaluation in conjunction with ophthalmologic and surgical evaluations. A multidisciplinary approach is required for the diagnosis and treatment of lesions in this area, which is an anatomic and functional entity. Longitudinal endocrine follow-up is also required to detect and correct any endocrine abnormalities associated with intracranial disease or arising as an unintended effect of treatment.
 Clinical Manifestations
Clinical Manifestations
Hormone Deficiency
Pituitary Hypersecretion
 Endocrine Evaluation
Endocrine Evaluation
Hormone Deficiency
Pituitary Hypersecretion
 Consequences of Endocrine Evaluation
Consequences of Endocrine Evaluation
 Conclusion
Conclusion
References
Related posts:
 Chemotherapy Options for Sellar and Parasellar Tumors
Surgical Treatment of Pituitary Adenomas
Stereotactic Radiosurgery
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Chemotherapy Options for Sellar and Parasellar Tumors
Surgical Treatment of Pituitary Adenomas
Stereotactic Radiosurgery
Radiologic Evaluation and Diagnosis for Pathology in the Sellar and Parasellar Region
Intracavitary Radiation for Cystic Craniopharyngiomas
Surgical Treatment of Chordomas and Chondrosarcomas
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


