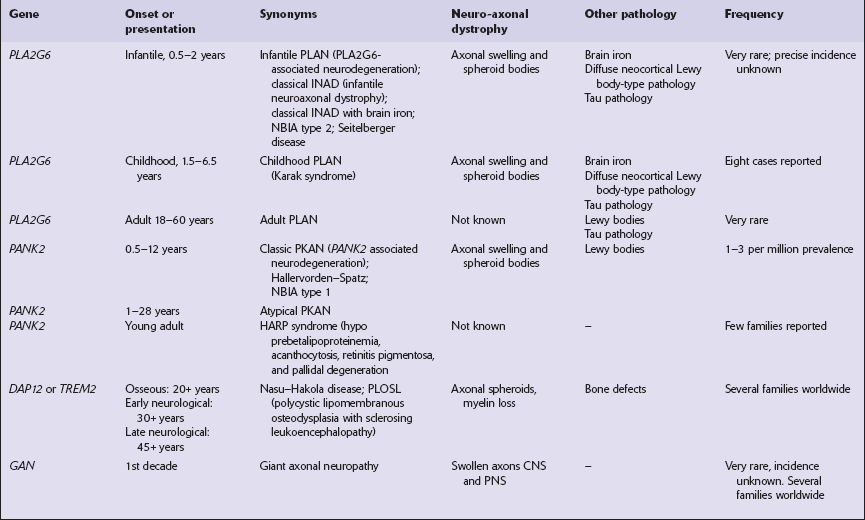
33 Thalamic degeneration is a feature of several multisystem neurodegenerative disorders (Table 33.1), but can also rarely occur in a ‘pure’ form. Table 33.1 Diseases in which thalamic degeneration may be prominent Multiple system atrophy (glial cytoplasmic inclusions) Spinocerebellar degenerations Wernicke’s encephalopathy Huntington’s disease Creutzfeldt–Jakob disease Menkes syndrome Membranous lipodystrophy Neuroaxonal dystrophy Fatal familial insomnia The grouping together of pallidal degenerations is based on the morphologic finding of degeneration centered on the globus pallidus, either alone or in combination with degeneration of the subthalamic nucleus or substantia nigra. These disorders have been subdivided according to the regions of the brain showing pathologic changes (Table 33.2). Clinically, pallidal degenerations are associated with a variety of movement disorders, with or without dementia. Some are familial and others sporadic. It is difficult to evaluate the nosologic status of many of the cases described in the literature as their relationship to disorders that can now be better defined by molecular genetic and immunohistochemical techniques is uncertain. In particular, cases of dentatorubropallidoluysian atrophy (DRPLA) (see Chapter 29), spinocerebellar atrophies (see Chapter 29), multiple system atrophy (see Chapter 28), and diseases characterized by the ubiquitinated inclusions seen in ALS (see Chapter 27) are probably included in most of the historic series. Once these entities are excluded, a group of pallidal degenerations remains that can be classified on a purely descriptive basis, although it is not clear to what extent they represent distinct diseases. Several conditions, grouped as neuroaxonal dystrophies, are characterized pathologically by the presence of axonal swellings, which are thought to develop as a result of neuronal dysfunction that produces distal axonal degeneration. In most cases, the nature of the neuronal dysfunction and the pathogenesis of the axonal swelling are poorly understood. Neuroaxonal dystrophy occurs in three contexts (Tables 33.3, 33.4)). Table 33.3 Classification of neuroaxonal dystrophic processes Physiologic neuroaxonal dystrophy: normal brain aging Gracile and cuneate nuclei Globus pallidus Substantia nigra Spinal anterior horns Primary neuroaxonal dystrophy: diseases in which the main pathology is neuroaxonal dystrophy Neuroaxonal dystrophies with PLA2G6 mutations (phospholipase A2 group VI) Infantile neuroaxonal dystrophy (INAD) Late infantile neuroaxonal dystrophy Juvenile neuroaxonal dystrophy Adult neuroaxonal dystrophy Childhood onset phospholipase A2 group 6-associated neurodegeneration (PLAN; atypical neuroaxonal dystrophy) Schindler disease (PLA2G6 mutation and co-occurrence of α-N-acetylgalactosaminidase (α-NAGA) deficiency) Neuroaxonal leukodystrophy Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) Familial pigmentary orthochromatic leukodystrophy (POLD) Pantothenate kinase-associated neurodegeneration (PKAN; formerly Hallervorden–Spatz disease) Classical PKAN Atypical PKAN Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome) Nasu–Hakola disease Giant axonal neuropathy (caused by mutation of GAN, the gigaxonin gene) Secondary neuroaxonal dystrophy: accentuation of neuroaxonal dystrophy in other disease processes Neurodegenerative disease Metabolic disease Infective disease Dystrophic axonal swellings can be identified in sections stained with hematoxylin and eosin as rounded or elongated eosinophilic structures varying from 20 μm to 120 μm in diameter (Fig. 33.1a,b). Some dystrophic axons contain an intensely stained eosinophilic core surrounded by a paler zone (Fig. 33.1c). The swollen axons can be demonstrated by silver impregnation techniques (Fig. 33.1d). In toluidine-blue-stained resin sections, the swellings are seen to contain granular material (Fig. 33.1e). Electron microscopy shows this to consist of mitochondria, electron-dense lysosome-related bodies, tubulomembranous structures, and amorphous matrix material (Fig. 33.1f). Generally, relatively few neurofilaments are present and those that are may be displaced towards the periphery of the axon. Immunoreactivities for neurofilament protein and ubiquitin are largely confined to axonal swellings smaller than 30 μm in diameter (Fig. 33.2). Iron-containing and lipofuscin-like pigment may accumulate in axonal spheroids (Fig. 33.3), leading to the descriptive term pigment-spheroidal dystrophy. 33.1 Microscopic features of neuroaxonal dystrophy. 33.2 Immunoreactivity of dystrophic axonal swellings. 33.4 Physiologic neuroaxonal dystrophy. MACROSCOPIC AND MICROSCOPIC APPEARANCES Axonal spheroids and reactive astrocytic gliosis are widely distributed in the central and peripheral nervous system (Fig. 33.5). Degeneration of the corticospinal and spinobulbar tracts is usually prominent. There is often a moderate to severe diffuse cortical Lewy body pathology as well as tau pathology with neurofibrillary tangles and neuropil threads. 33.5 Infantile neuroaxonal dystrophy.
Thalamic and pallidal degenerations, neuroaxonal dystrophy, and autonomic failure
THALAMIC DEGENERATIONS
PURE THALAMIC ATROPHY
 DIFFERENTIAL DIAGNOSIS OF PURE THALAMIC ATROPHY
DIFFERENTIAL DIAGNOSIS OF PURE THALAMIC ATROPHY
 The conditions listed in Table 33.1 should be excluded. These include fatal familial insomnia, which has a primary thalamic pattern of involvement and is a form of prion disease (see Chapter 32).
The conditions listed in Table 33.1 should be excluded. These include fatal familial insomnia, which has a primary thalamic pattern of involvement and is a form of prion disease (see Chapter 32).
 Neuronal loss from the thalamus has been described in patients that have undergone leukotomy.
Neuronal loss from the thalamus has been described in patients that have undergone leukotomy.
PALLIDAL DEGENERATIONS
NEUROAXONAL DYSTROPHY
MICROSCOPIC APPEARANCES
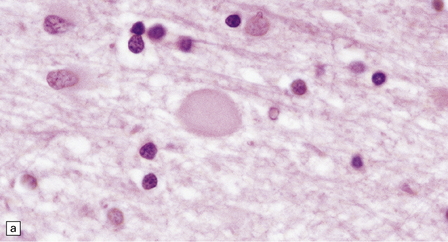
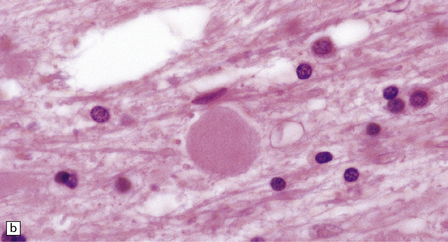
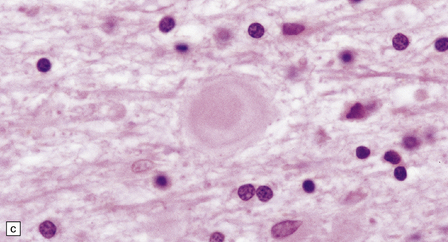
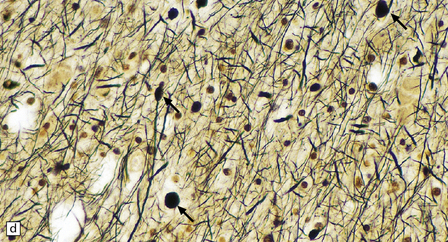
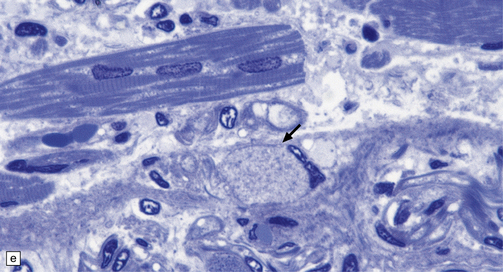
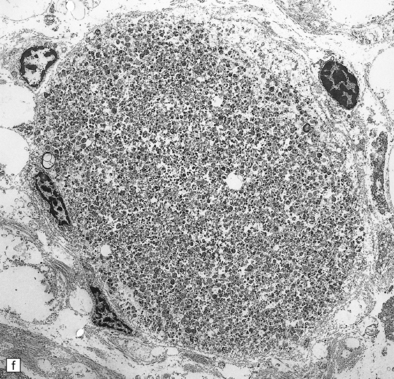
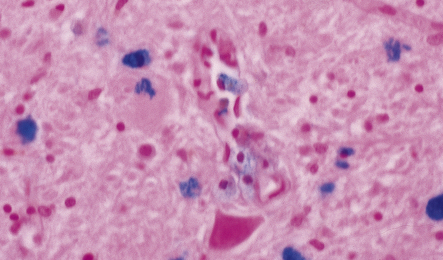
(a) Axonal spheroids appear as pale eosinophilic rounded structures. (b) Many have a granular appearance in sections stained with hematoxylin and eosin. (c) Some spheroids show slight central pallor. Others, particularly the large spheroids, may contain a densely eosinophilic central region and a paler surrounding zone, as shown here. (d) The axonal swellings can be demonstrated by silver impregnation (arrows). (e) In resin sections, spheroids (arrow) appear pale and granular. (f) Electron microscopy shows that the spheroids contain abundant electron dense granules, mostly membrane-bound, and derived from lysosomes and degenerating mitochondria.
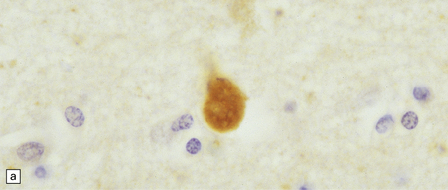
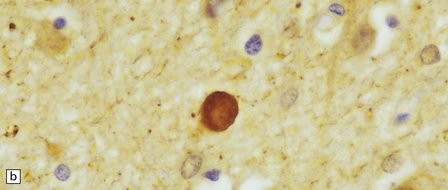
(a) Axonal swellings can be detected by immunohistochemistry for ubiquitin. (b) They can also be detected by immunohistochemistry for neurofilament protein. Strong immunoreactivity is generally seen only in small spheroids.
PHYSIOLOGIC NEUROAXONAL DYSTROPHY
 Gracile and cuneate nuclei in the medulla (Fig. 33.4a).
Gracile and cuneate nuclei in the medulla (Fig. 33.4a).
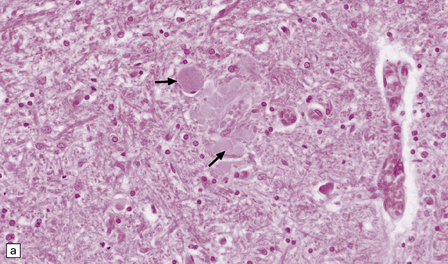
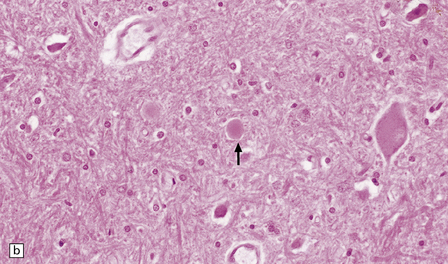
(a) Pale eosinophilic axonal swellings (arrows) in the gracile nucleus of an elderly person. (b) In the substantia nigra, axonal swellings (arrow) are typically more eosinophilic.
 Zona reticulata of the substantia nigra (Fig. 33.4b).
Zona reticulata of the substantia nigra (Fig. 33.4b).
PRIMARY NEUROAXONAL DYSTROPHY
 Diseases with PLA2G6 mutations: infantile, late infantile, juvenile, and adult neuroaxonal dystrophy.
Diseases with PLA2G6 mutations: infantile, late infantile, juvenile, and adult neuroaxonal dystrophy.
NEUROAXONAL DYSTROPHY WITH PLA2G6 Mutations
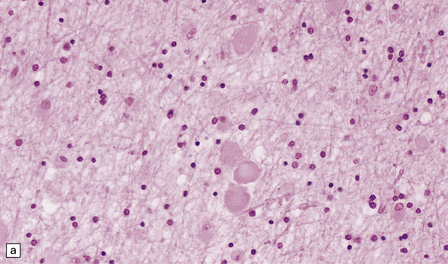
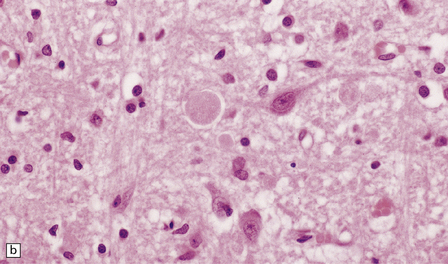
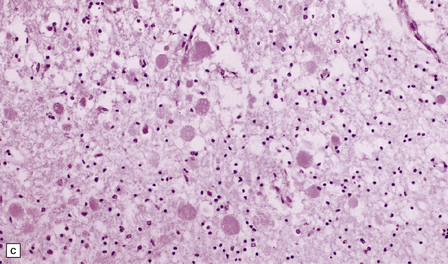
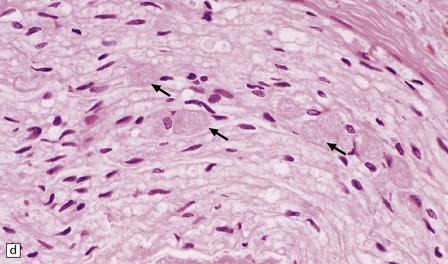
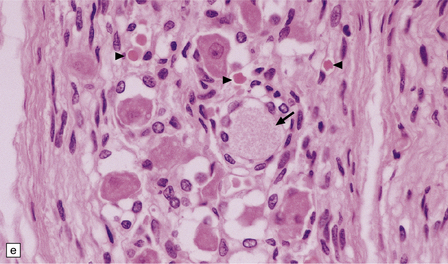
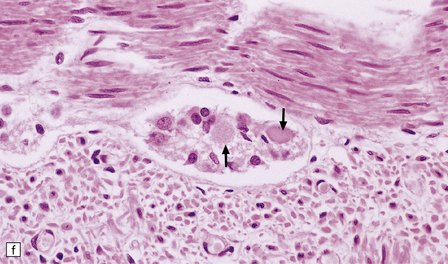
(a) Axonal spheroids are widely distributed in both white and gray matter in the CNS, as shown here in the hemispheric white matter. (b) Axonal spheroids in the basal ganglia. (c) Axonal spheroids are especially prominent in long tracts in the spinal cord. (d) Dystrophic axonal swellings (arrows) composed of granular eosinophilic material are also seen in peripheral nerves. (e) Dystrophic axonal swellings (arrowheads) and a degenerating neuron (arrow) in sympathetic ganglia. (f) Dystrophic axonal swellings (arrows) in the myenteric plexus of the gut. In the CNS, the formation of spheroids is followed by loss of myelin, neuronal degeneration, and astrocytic gliosis.![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thalamic and pallidal degenerations, neuroaxonal dystrophy, and autonomic failure
 PURE THALAMIC ATROPHY
PURE THALAMIC ATROPHY


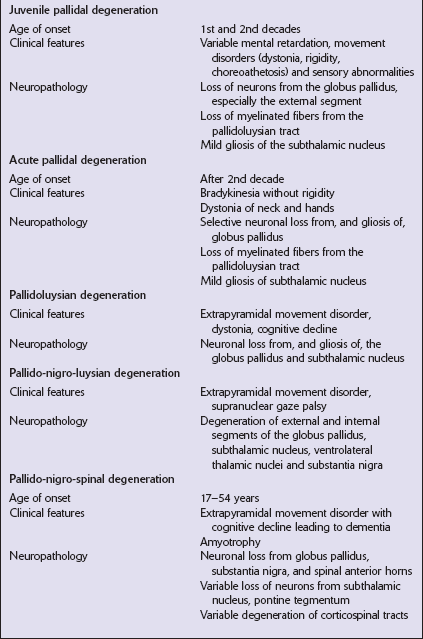
 DIFFERENTIAL DIAGNOSIS OF PALLIDAL DEGENERATIONS
DIFFERENTIAL DIAGNOSIS OF PALLIDAL DEGENERATIONS






Only gold members can continue reading. Log In or Register to continue


