Chapter 10 The EEG in Epilepsy
SEIZURE TYPES AND SEIZURE SYNDROME
Seizure Types
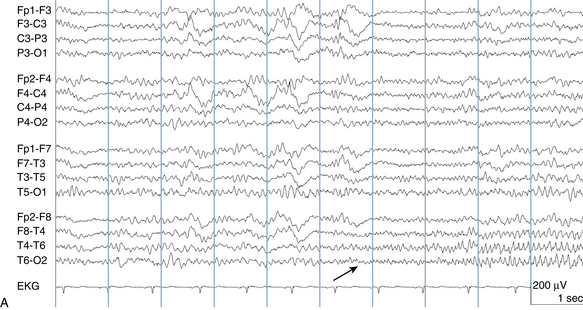
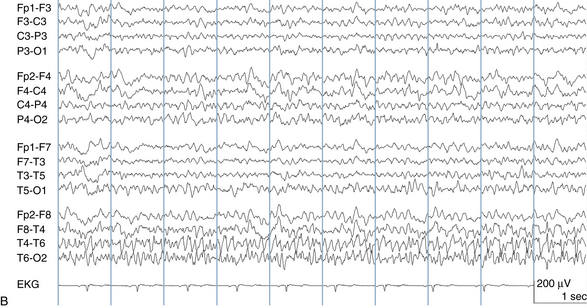
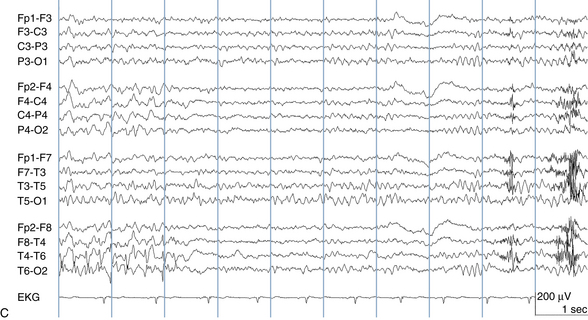
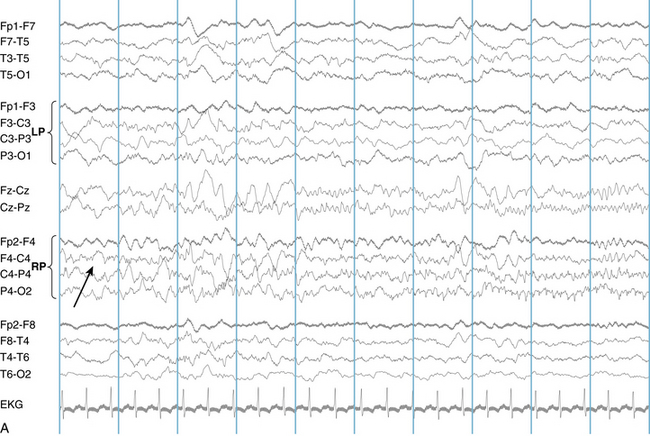
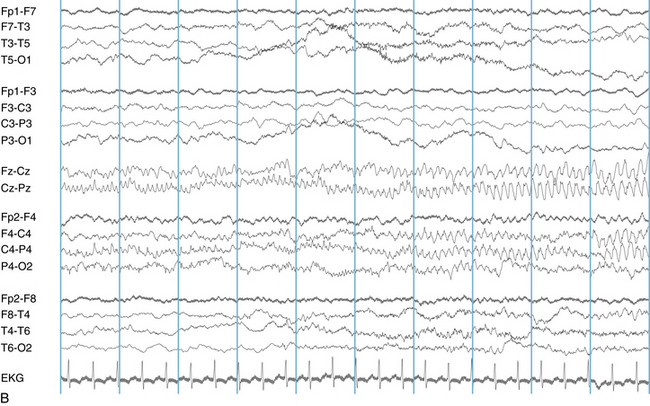
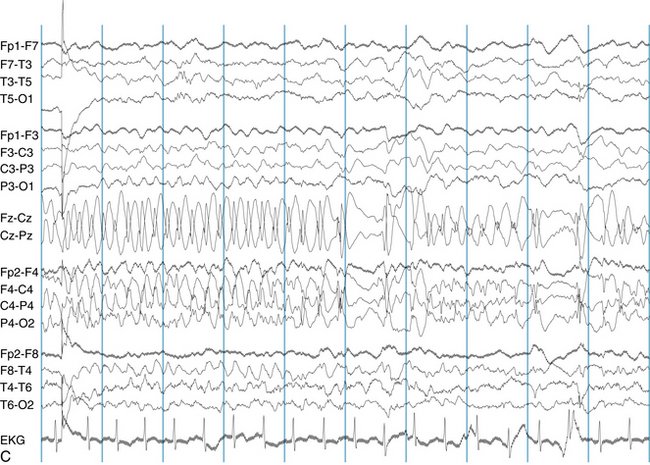
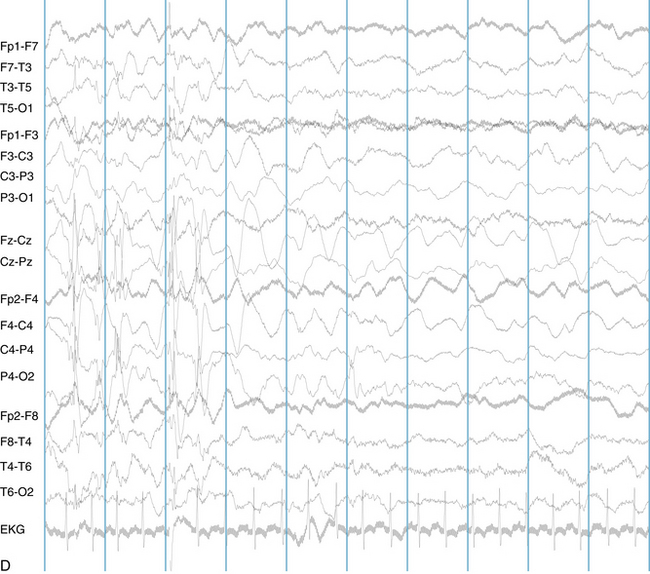
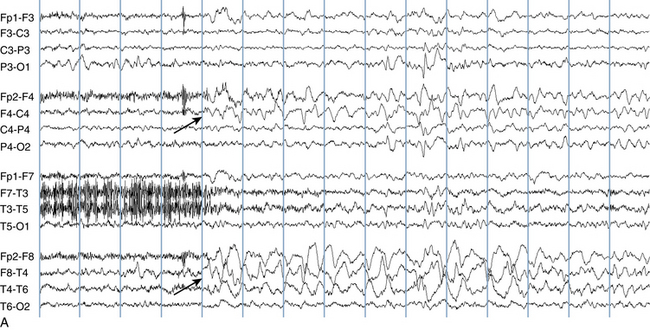
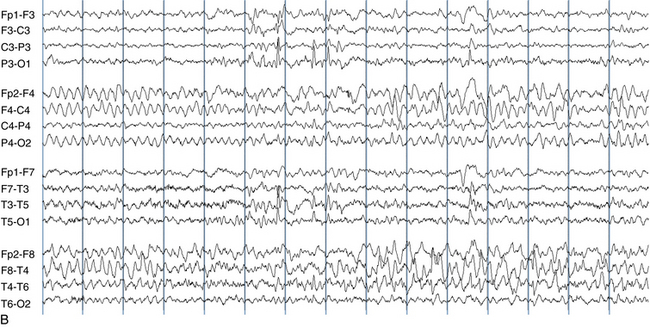
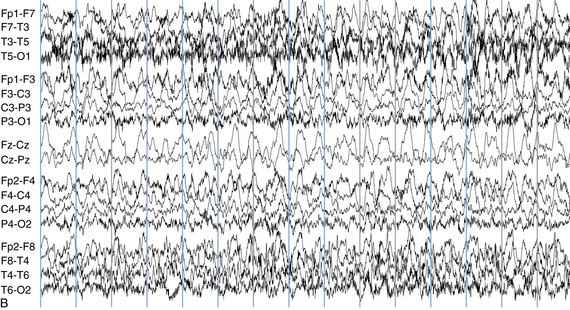
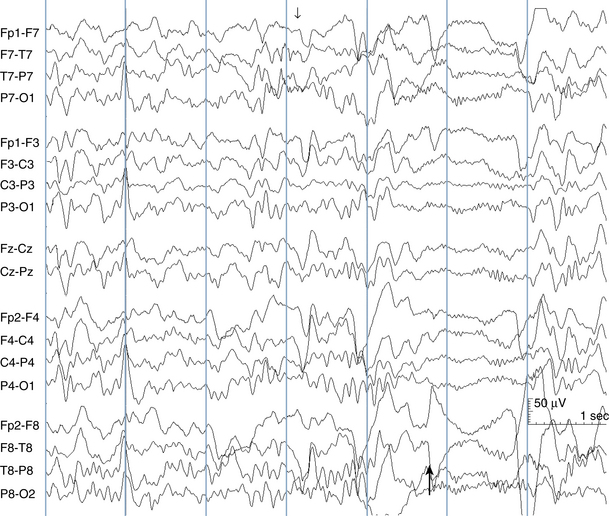
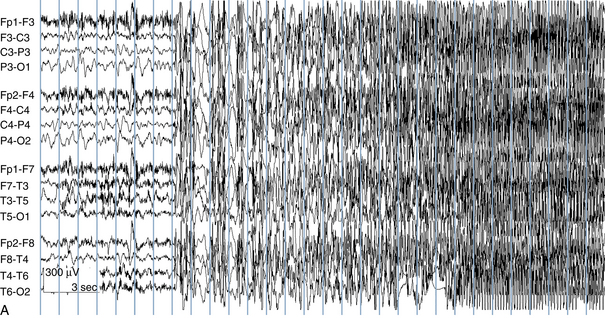
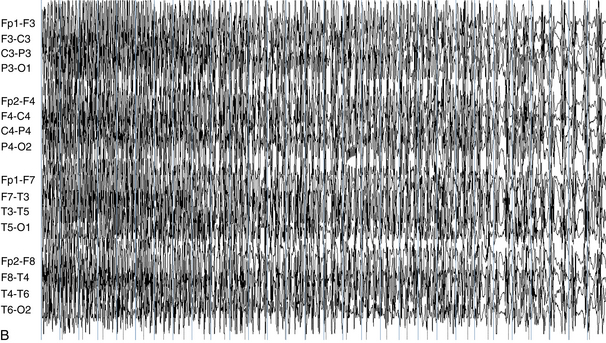
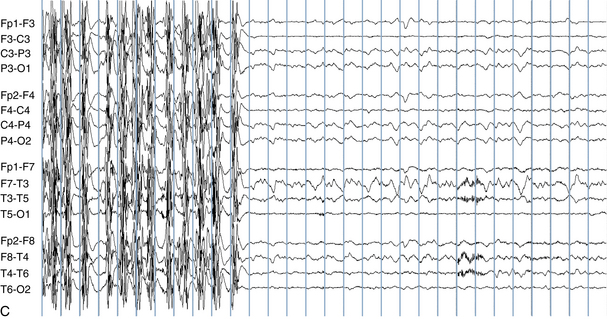
The following hypothetical example illustrates how these three key features are used: a patient experiences an event that starts with a subjective report of a feeling of fear. Next, observers report that the patient begins to stare and is unresponsive, followed by rhythmic jerking of the left arm. Simultaneously, an EEG is recorded that shows a rhythmic discharge starting in the right temporal lobe and evolving to include much of the right hemisphere over a brief period of time (see Figure 10-1, A, B, and C). The combination of psychic aura reported by the patient, staring, the specific motor phenomena reported by observers, and observation of an EEG seizure discharge that starts in the right temporal lobe all establish the diagnosis of a specific seizure type: a complex partial seizure arising from the right temporal lobe. Note that the age and previous history of the patient are not of primary importance in diagnosing seizure type; rather, the behaviors observed during the episode and any recordings made at the time of the event are the most important elements in defining seizure type. We can assign the seizure type “complex partial seizure arising from the right temporal lobe” or “right temporal lobe seizure” without knowing whether the seizures are occurring in a broader context of possible posttraumatic epilepsy or cryptogenic temporal lobe epilepsy. This contrasts with the approach to diagnosing seizure syndromes, described next.
CLASSIFICATION OF SEIZURE TYPES
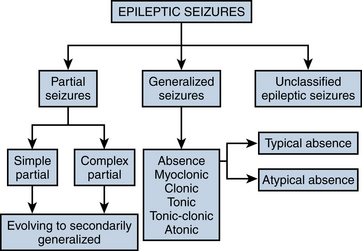
The most frequently used classification of seizure types was established by a committee of the International League Against Epilepsy (ILAE) in 1981. This classification divides seizures into partial and generalized categories and a smaller unclassified category. The process of splitting seizure types up into generalized or partial categories is the first step in seizure classification. Each category of seizure type is described briefly in this section, along with its characteristic clinical and EEG findings. The broad schema of the classification is shown in Figure 10-2.
Partial Seizures
Because a seizure can arise from nearly any location on the cortical surface, the range of potential seizure manifestations is quite diverse. Partial seizures may involve motor findings (such as jerking or stiffening of a limb), sensory findings (such as a sensation of tingling or pain over a region of the body, hearing a sound, smelling an odor, or seeing brightly colored shapes), psychic features (such as a sensation of fear or déjà vu), or autonomic findings (such as sweating or palpitations). It should also be kept in mind that a significant portion of the cortical surface is functionally relatively silent. When a seizure discharge starts in one of these “silent” cortical areas, it is possible for the discharge to occur on the cortical surface without clinical change as a clinically silent seizure rather than a clinical seizure. Frequently, a seizure discharge may begin in one of these “silent” areas and subsequently propagate to other areas such as the motor strip, at which time obvious clinical signs may appear such as clonic jerking of an extremity. Thus, when a patient manifests clonic jerking, we can infer that the seizure discharge has involved the motor strip, but this is not a guarantee that the seizure discharge originated in the motor strip—the seizure discharge may have started elsewhere and propagated to the motor strip. Figures 10-3 and 10-4, show examples of how two focal-onset seizures propagate.
Recordable EEG Patterns Associated With Partial Seizures
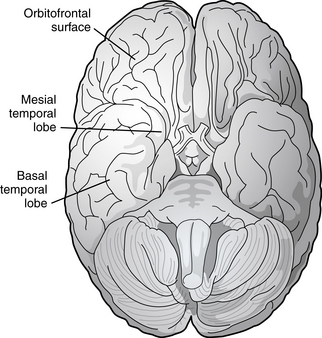
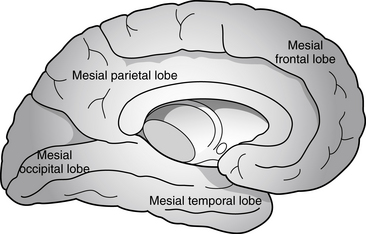
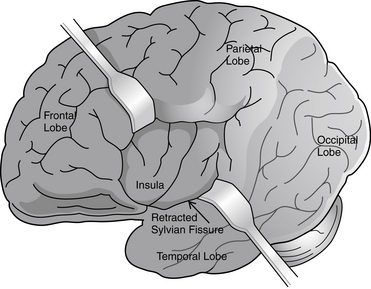
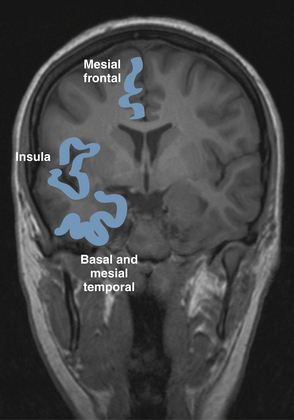
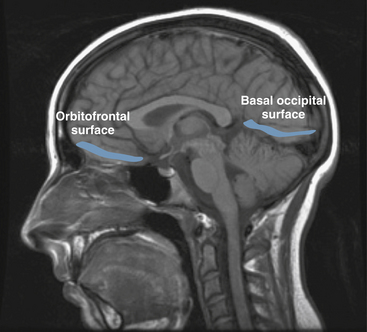
Partial seizures are distinguished by EEG patterns that involve only subsets of the brain. In comparison, the EEG patterns of generalized seizures involve all brain areas at once. Although most epileptic seizures can be recorded using standard scalp electrodes, in some cases a seizure discharge may occur in a cortical area that is not readily accessible to recording with conventional electrodes. These include such areas as the mesial surfaces of the frontal, parietal, and occipital lobes, the orbitofrontal surface of the frontal lobe, the basal occipital and temporal lobes, and the mesial surface of the temporal lobe and insula, among others (see Figures 10-5 through 10-9). Therefore, most, but not all partial seizures are well recorded at the scalp. For these reasons, a negative EEG recording cannot, in and of itself, exclude the diagnosis of an epileptic seizure. In these occasional cases of definite epileptic seizure with negative scalp recordings, other factors must be taken into account to make the correct diagnosis, such as the specific features of the patient event and the history. In cases of partial epileptic seizures associated with a negative EEG, the definition implies that there is some theoretical electrode placement that could record the seizure discharge, even if that placement location would have to be deep within the brain.
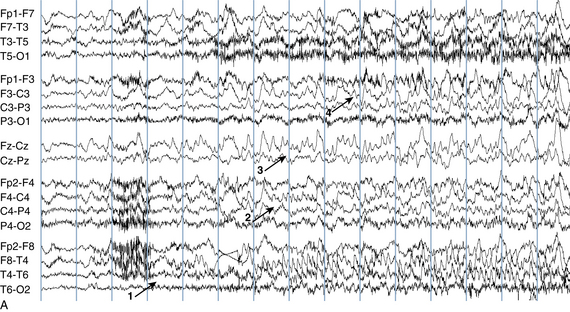
The EEG patterns associated with partial seizures usually consist of a rhythmic, sharp discharge over the affected area (e.g., spike or spike-wave discharges) as shown in the previous figures. However, partial seizure recordings do not only appear as a train of spikes. Especially when seizure sources are located deeper in the brain and at some distance from the recording electrode, the seizure may only appear as a rhythmic focal slow wave without obvious sharp features when recorded from the scalp. In the case of partial seizures, the epileptic discharge is usually unilateral. Although a unilateral partial seizure discharge may spread incrementally through the primary involved hemisphere, at such time as the discharge might cross to the opposite hemisphere, both hemispheres become simultaneously engulfed with seizure activity; there is no incremental spread through the opposite hemisphere. This process is referred to as secondary generalization.
An important exception to this rule is the example of temporal lobe seizures in which the seizure discharge may spread from one temporal lobe to the other without simultaneous involvement of the remainder of the hemispheres (see Figure 10-10). Therefore, in most cases, after the discharge becomes bilateral, the whole of both hemispheres is engulfed with the discharge and the discharge can be considered to have generalized. Complex partial seizures with bilateral temporal involvement represent the exception to this rule: the less typical example of a bilateral seizure discharge that is not truly generalized.
Partial Seizures With Secondary Generalization
As described earlier, a seizure discharge may start focally and subsequently spread to involve all brain areas (generalize). This flow of the discharge from a subset of cerebral cortex to all of cerebral cortex is reflected both by the spread of the recorded discharge from a subset of EEG channels to all EEG channels and also by an evolution of the patient’s seizure behavior from a partial manifestation to involvement of the whole body. For instance, in the classic example of the type of seizure referred to as a “Jacksonian march,” a patient’s seizure may begin with clonic contractions in the right hand and arm and subsequently spread to the right face and leg. Thereafter, it may spread to the opposite side of the body so that bilaterally synchronous clonic activity of the whole body is seen. This clinical progression is mirrored by an electrographic evolution of the discharge from a small area in the left hemisphere, which includes the portion of the motor strip that is associated with the left hand, to the whole of the left hemisphere and then, finally, to involvement of both hemispheres (see Figure 10-11).
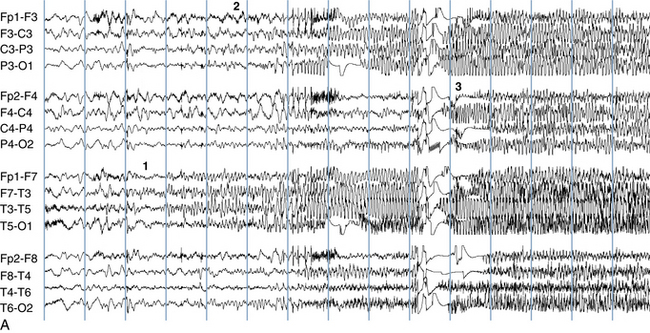
Partial seizures that secondarily generalize are classified among the partial seizures rather than the generalized seizures for diagnostic reasons. Partial seizures that do not generalize and partial seizures that do secondarily generalize have the same list of possible causes. In comparison, the list of causes of generalized seizures is distinctly different from the list of causes of partial-onset seizures. The tendency to a partial seizure to secondarily generalize usually has little to do with the etiology of the seizure.
Typical Absence Seizures
EEG
The most frequent EEG correlate to typical absence seizures is the “classic” 3-Hz generalized spike-wave discharge (see Figure 10-12). When these discharges are analyzed closely, the maximum voltage of the spike component of the spike-wave complexes is most commonly seen in the superior frontal electrodes (F3 and F4). Less often, the spike maximum is seen in the occipital area, and even less frequently in other locations.
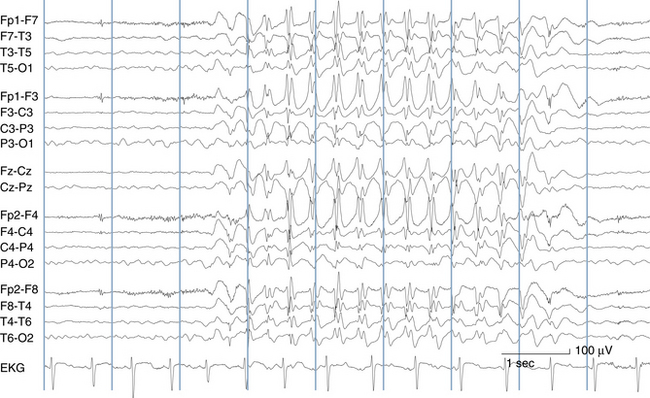
Although the term 3-Hz generalized spike wave is well known and implies a consistent frequency, observed firing frequencies are not necessarily as consistent as the term implies. Often, the first few discharges fire at a frequency slightly faster than 3 Hz. After onset of the discharge, the firing frequency typically slows, often to 2.5 Hz and sometimes to 2 Hz before abruptly terminating (see Figure 10-13). One of the most characteristic attributes of the typical absence seizure is the abrupt onset and termination of the discharge. The classic 3-Hz generalized spike-wave discharge tends to occur against a normal background and has a clear time of onset and a fairly well demarcated termination. After termination, the EEG returns to the previous background after a few seconds or less.
Atypical Absence Seizures
As with typical absence seizures, the main clinical features of atypical absence seizures are staring and unresponsiveness. Atypical absence seizures differ, however, in that onset and termination of the episodes, both clinically and electrographically, are less clear, and firing rates are slower. Also, atypical absence seizures tend to occur in individuals with cognitive impairment or mental retardation, whereas typical absence seizures are more often seen in subjects who are cognitively normal. Changes in tone are more common during atypical absence seizures, with slumping of the head, shoulders, and sometimes the whole torso seen during some examples.
EEG
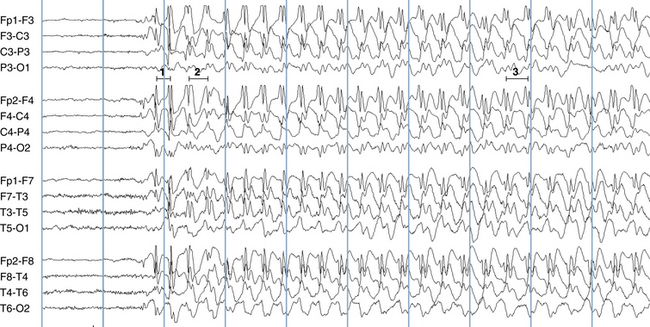
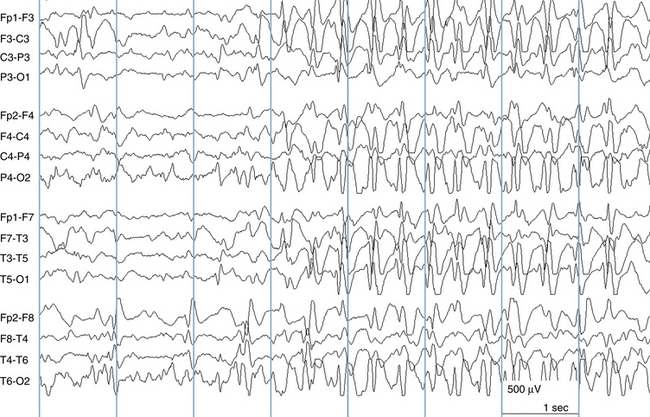
The EEG hallmark of atypical absence seizures is the slow spike-wave discharge. Slow spike-wave discharges differ from “classic” 3-Hz generalized spike-wave discharges in two important respects: slow spike-wave discharges, as their name implies, fire at a slower rate, usually 2.5 Hz or less at onset; see Figure 10-14). Slow spike-wave discharges also lack the clear-cut onset and termination characteristic of typical absence seizure discharges. Finally, whereas slow spike-wave discharges are often generalized, asymmetries, both between the left and the right hemispheres and the anterior and posterior head regions, are more common. There is no single characteristic location for the discharge maximum for the slow spike-wave discharges associated with atypical absence seizures, and the location of the voltage maximum may differ even within the same patient at different times. Atypical absence seizures often occur against the backdrop of an otherwise abnormal EEG, which may include scattered epileptiform activity or a slowed background.
Myoclonic Seizures
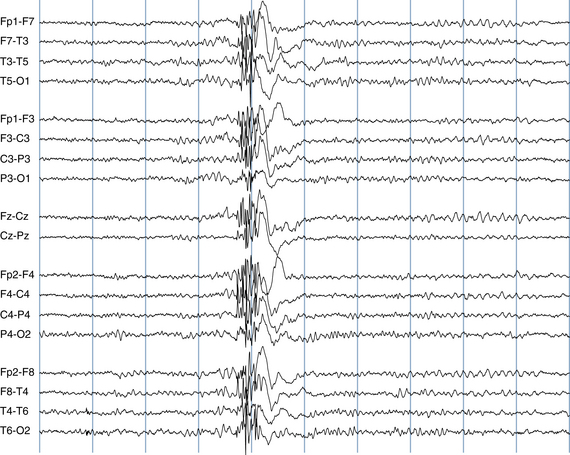
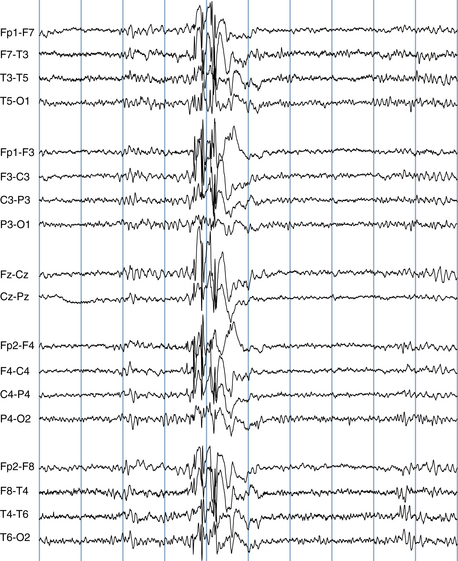
Myoclonic seizures consist of a lightning-like or shock-like contraction of the muscles driven by an epileptic discharge. The appearance of a myoclonic seizure is similar to experiencing a brief electric shock. Myoclonus may consist of a single jerk or a quick series of jerks that occur in a burst, either rhythmic or nonrhythmic. Epileptic myoclonus may manifest as a muscle jerk in nearly any part of the body, although the most common location for epileptic myoclonus is the upper shoulder girdle. In such cases, the myoclonus usually consists of a quick series of abduction jerks at the shoulders. During the series of jerks, there may be a tendency for slight net abduction of the upper arms away from the body with each jerk. The most common EEG manifestation of epileptic myoclonus is a high-voltage polyspike-wave discharge, which may occur singly or in brief, repetitive bursts (see Figures 10-15 and 10-16).
Clonic Seizures, Tonic Seizures, and Generalized Tonic-Clonic Seizures
Tonic Seizures
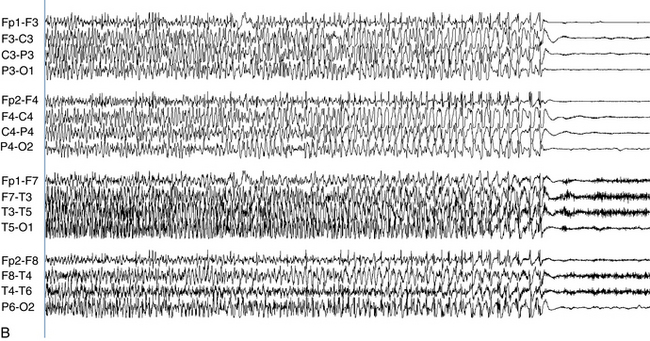
Tonic seizures cause tonic stiffening of a limb, several limbs, or the whole body. When the whole body is involved with a generalized tonic seizure, tonic stiffening in extension is most common; however, tonic stiffening with flexion of the hips, knees, or arms may also be seen. Especially with generalized tonic seizures, tonic contraction of the diaphragm may occur resulting in a forced inhalation or grunting sounds. Most often the EEG correlate of tonic seizures is a spray of rapid spikes in the affected area, often with an initial frequency of 10 to 25 Hz, which subsequently slows, similar to the onset of tonic-clonic seizures as described next. Other EEG correlates are not uncommon, including an abrupt desynchronization (flattening) of the EEG. When such flattening is seen, there is sometimes a suggestion of low voltage rapid spikes superimposed on the flattened pattern though these are not always identifiable (see Figure 10-17). Therefore, although most ictal patterns are dramatic and show high-voltage repetitive discharges, the electroencephalographer must also be alert to abrupt flattening of the EEG (an electrodecrement) as a seizure correlate. Other patterns are less common.
Generalized Tonic-Clonic Seizures
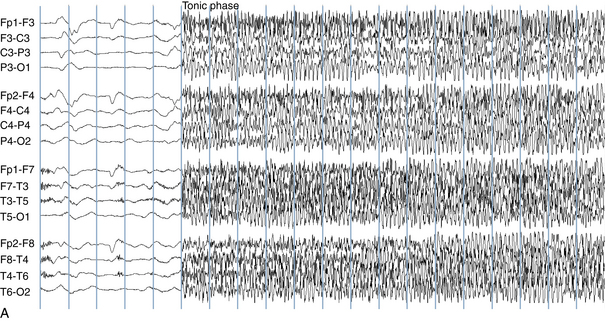
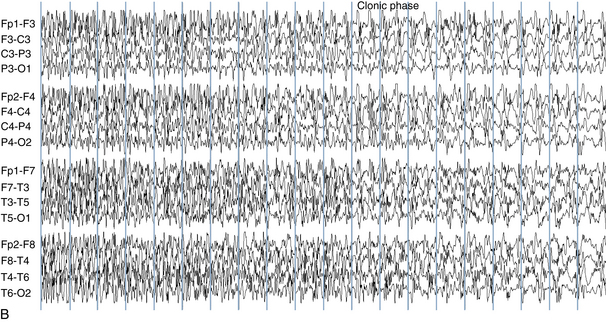
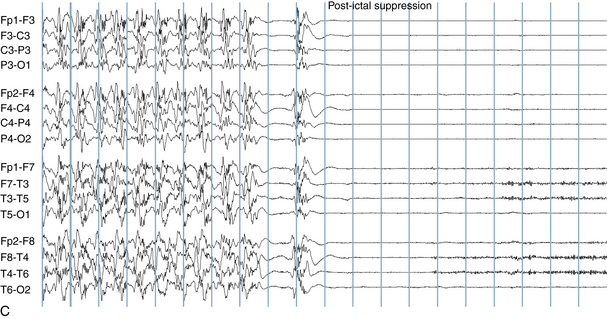
The EEG correlate of the tonic-clonic seizure often begins with an abrupt onset of generalized rapid spikes which then slow in frequency over the course of the event. As the firing frequency of the spikes slows, the spikes may begin to manifest a clearer spike-wave morphology. From the clinical perspective, rapid spikes are often associated with tonic stiffening. At a certain point in the seizure, the firing rate of the spikes slows to a point that allows each spike or spike-wave discharge to generate a separate clonic jerk (see Figure 10-18). This is the reason that the progression from tonic stiffening to clonic jerking is so common. Over the course of a generalized tonic-clonic seizure the clonic jerking is seen to slow in frequency and blend into a slow-wave pattern: “postictal slowing.” Less commonly, a clonic-tonic-clonic seizure may occur in which clonic jerking speeds up and melds into tonic stiffening, followed by the usual progression back to clonic jerking. As expected, the EEG correlate of this type of seizure often consists of spike-wave discharges that speed up to become rapid spikes, and then slow down again (see Figure 10-19).
Neonatal Seizures
Neonatal seizures have been given a classification scheme different from the general ILAE seizure classification described earlier. The most common neonatal seizure classification system in use today was described by Volpe in 1989. The seizure types described in this classification are actually quite similar to those of the ILAE classification, with the exception of an additional seizure category referred to as “subtle seizures,” a subgroup that remains controversial as the epileptic nature of subtle seizure behaviors has not yet been firmly established. See Chapter 13, “The EEG of the Newborn” for further discussion.
CLASSIFICATION OF SEIZURE SYNDROMES
Epileptic Syndromes of Early Infancy Associated With a Burst-Suppression Pattern
Early Myoclonic Encephalopathy
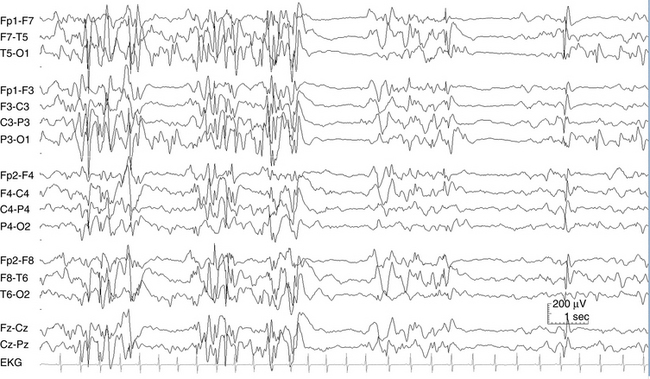
Infants with EME present soon after birth with both fragmentary and massive myoclonic seizures. Other seizure types also occur. The magnetic resonance imaging (MRI) scan at birth is almost always normal, and a large fraction of these babies are eventually found to have a specific metabolic disorder, nonketotic hyperglycinemia (NKH). Those babies with EME who do not prove to have NKH may have other metabolic diagnoses, and it is felt that the remaining cases of EME may be caused by some metabolic entities yet to be defined. This belief is based on the observations that EME babies have anatomically normal brains by MRI, and there is usually no history of a previous neurological injury. Babies with EME have complete developmental failure, and the seizures tend to be refractory to treatment. The EEG shows an unremitting burst-suppression pattern that may continue unabated through childhood (see Figure 10-20).
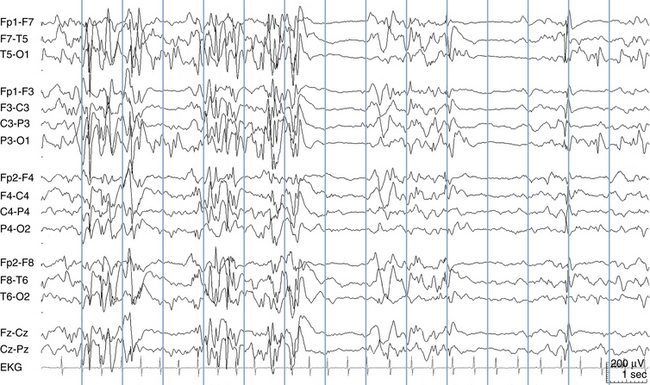
Early Infantile Epileptic Encephalopathy
Like EME, EIEE is typically associated with a burst-suppression pattern on EEG. The EEG patterns of EIEE and EME are not easily distinguished without the benefit of the clinical history (see Figure 10-21). The burst-suppression pattern of EIEE is more likely to evolve into other EEG background patterns later in life, compared with the EME burst-suppression pattern, which may persist indefinitely.
The Concept of the Age-Dependent Epileptic Encephalopathies
EIEE is considered one of the age-dependent epileptic encephalopathies. This group of syndromes represents the result of different characteristic epileptic responses of the brain to injury seen at different ages. For instance, when the brain is injured in the neonatal period, the response to injury may present as the EIEE syndrome as described earlier. When the epileptic response to an injury or abnormality occurs later in infancy (typically after 3 months of age), the child may develop a pattern of infantile spasms or West syndrome (discussed later). The epileptic response to an injury that appears after 3 years of age may present as the Lennox-Gastaut syndrome (discussed later). Because different maturational states of the brain are only associated with certain syndromic patterns, West syndrome does not present in adults, and Lennox-Gastaut syndrome cannot present in early infancy; because of the way the human brain matures, it is not able to mount those types of seizure patterns at those ages.
Benign Familial Neonatal Convulsions and Benign Neonatal Convulsions
Benign Neonatal Convulsions
The family history for seizures is negative. There is no antecedent history of a difficult delivery or birth injury, and neuroimaging is normal. Apart from possible mild hypotonia, the interictal examination is normal. A search for central nervous system infection is negative, and no electrolyte or other metabolic disturbances are found. The clinician is left with a story of an otherwise perfectly well newborn with unexplained onset of seizures in whom all testing is normal.
In babies with benign neonatal convulsions, the EEG background pattern tends to be normal. A characteristic EEG finding has been described in such babies, termed théta pointu alternant. This is a pattern of sharpened theta waves occurring in brief runs, typically in each central area, and alternating sides (see Figure 10-22). Although this pattern is said to occur in the majority of patients with this seizure syndrome, it may be difficult to identify, and its presence is not necessarily diagnostic of benign neonatal convulsions.

(Modified from Dehan M, Quillerou D, Navelet Y, et al. Convulsions in the fifth day of life: a new syndrome? Arch Fr Pediatr 1977;34:730 742).
Febrile Seizures
A relationship between febrile seizures and temporal lobe epilepsy has long been suspected. Case-control studies of individuals with temporal lobe epilepsy appear to show an increased incidence of a history of febrile seizures, particularly prolonged febrile seizures, in temporal lobe epilepsy patients compared with control subjects. These findings suggest the possibility that prolonged febrile seizures may cause hippocampal damage (hippocampal sclerosis) and predispose to later temporal lobe epilepsy. An alternative interpretation is that those children destined to have temporal lobe epilepsy later in life may automatically be more prone to prolonged febrile seizures in childhood and that the early febrile seizures may not be a causative factor in the later epilepsy.
Infantile Spasms and West Syndrome
EEG in the Diagnosis of Infantile Spasms and West Syndrome
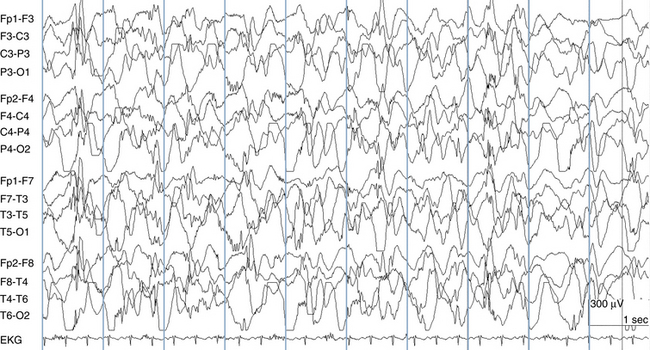
The EEG term hypsarrhythmia is derived from the Greek meaning “high” or “lofty” rhythm. In fact, some of the highest voltages measured in electroencephalography are seen in babies with hypsarrhythmia. Compared with adult EEGs in which voltages typically do not exceed 200 μV, hypsarrhythmia EEGs may exceed 1 mV (1000 μV). The essential features of the hypsarrhythmic pattern are high voltage and chaos. In this context, chaos refers to the opposite of synchrony. A completely chaotic EEG pattern is a pattern in which different electrical events and rhythms are occurring in different brain regions at different times in an unsynchronized and seemingly unrelated fashion. In contrast, the generalized spike-wave discharge, although abnormal, represents a pattern with a high degree of synchrony with all cortical areas acting in unison. In the chaotic hypsarrhythmia pattern, rapid spikes, high-voltage s low waves, and other abnormalities may occur in scattered locations at different times and in a seemingly random fashion (see Figure 10-23). Intermediate states between synchrony and complete chaos are also seen (see Figure 10-24).





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


