INTRODUCTION
The frontotemporal dementia (FTD) syndromes are related disorders resulting from neurodegeneration of the frontal and anterior temporal lobes. In 1892, Arnold Pick, a Prague neuropsychiatrist, described the first known patient and subsequently published a series of three patients with left anterior temporal atrophy1,2. In 1911, Alois Alzheimer, a German neuropathologist, described the characteristic inclusion bodies of what came to be known as ‘Pick’s disease.’ The field was nearly quiescent until the late 1980s when investigators in Lund, Sweden reported on an autopsy series of 158 patients with dementia3‘4, 20 (13%) of whom proved to have frontal lobe degeneration. In the past two to three decades, progress has been rapid. In the 1990s, there were several clinicopathological studies of FTD5,6 and the eventual demonstration of a linkage of some cases to the tau gene region on chromosome 177. Finally, in the past few years, researchers discovered an association of FTD with mutations in the progranulin gene (PGRN) and identified one of the major ubiquitinated proteins in FTD as the TAR DNA-binding protein 43 (TDP-43)8-11.
The three principal FTD syndromes are the behavioural variant (bvFTD), which comprises over half of these patients, and the language-predominant syndromes of progressive non-fluent aphasia (PNFA) and semantic dementia (SD). Although often underdiagnosed12,13, these three FTD syndromes comprise the third most common neurodegenerative dementia after Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB). Clinical reports describe frequencies of FTD of 5-13% among dementia outpatients14,15. In possibly the best population-based series, there was a calculated annual (1990-1994) incidence of FTD in Rochester, MN, of 2.2 per 100 000 in the age group 40-49 years, 3.3 for 50-59, and 8.9 for 60-6915.
On average, the FTD syndromes have a presenile age of onset, are equally common among men and women, and lack a proven ethnic predisposition. The age of onset of bvFTD averages around 57 years with a usual range of 51 -63 years, but patients have presented from the 20s to the 80s16-18. The age of onset of SD is similar to that of bvFTD, but PNFA patients are often slightly older17. FTD syndromes are common among dementia patients with an age of onset of less than 65 years, accounting for approximately 15-20% of young-onset dementias18-20, with prevalences in the 45-64 year age group of 15 per 100 000 in the UK and 6.7 per 100 000 in the Netherlands20-22.
CLINICAL PRESENTATIONS Behavioural Variant FTD
The clinical syndrome of FTD presents with several ‘core’ and ‘supportive’ behavioural changes in mid-life (Table 57.1)23 – 26. The most common behavioural changes are apathy or decreased spontaneity and interest, and disinhibition or impulsivity25,27,28. Second, bvFTD patients violate social and moral norms, conventions or rules25,26. They may inappropriately touch others or violate their personal space, talk to strangers or children that they do not know, make sexual comments, forgo tact or table manners, and even manifest sociopathic behaviour such as theft. Third, they have ‘emotional blunting’ with a loss of empathy and awareness of the needs of others. Basic emotional appreciation is decreased, particularly for negative facial or vocal emotions29,30. Fourth, patients lose insight into their behavioural changes and lack self-referential behaviours such as embarrassability or shame. Fifth, they frequently have changes in dietary or eating behaviour, most commonly a carbohydrate craving25. Sixth, bvFTD patients have a range of repetitive behaviours from simple motor stereotypies to complex compulsive-like acts25. Finally, a supportive criterion is neglect or loss of interest in personal hygiene with failure to wash, bathe, groom or dress appropriately. In comparison, psychotic symptoms such as delusions and hallucinations are much less common among bvFTD patients compared to other dementia syndromes25,31.
Cognitive functions
Patients with bvFTD develop frontal-executive deficits. This is reflected in their lack of insight and loss of awareness or distress at their disability or the consequences of their behaviour. Their judgement is abnormal, and they are often concrete on idiom and proverb interpretation. Occasionally, patients have stimulus-bound behaviour such as echolalia and utilization behaviour (grasping and repeatedly using objects that they see or tending to read aloud anything they see). On language examination, bvFTD patients often have progressively decreased verbal output and mutism in advanced stages. They also manifest reiterative speech or verbal stereotypies. In contrast, early bvFTD patients have relatively spared memory with better free recall, cued recall and recognition than do AD patients18,32,33. Furthermore, they have preserved visuo- spatial skills such as spatial localization and orientation in familiar surroundings34,35. Rare bvFTD patients even have a facilitation of artistic and musical ability36.
Table 57.1 Behavioural variant frontotemporal dementia
Source: Modified from Neary et al.6
| I. Core diagnostic features |
| A. Progressive deterioration of behaviour, cognition or both |
| B. Early apathy or inertia |
| C. Early behavioural disinhibition with abnormal sociomoral behaviour |
| D. Early ‘emotional blunting’ or loss of sympathy or empathy |
| E. Loss insight or and self-referential behaviours |
| II. Supportive diagnostic features |
| A. Dietary or eating behaviour, most commonly a carbohydrate craving |
| B. Repetitive (stereotypic or compulsive) behaviour |
| C. Executive deficits with relative sparing of memory and visuo-spatial functions |
| D. Decline in personal hygiene and grooming |
| E. Brain imaging: predominant frontal and/or anterior temporal abnormality |
On neurological examination, bvFTD patients are usually normal early in their course but can develop primitive reflexes and motor changes as the disease progresses. A major subgroup of bvFTD patients develops parkinsonism, dystonia and ideomotor apraxia, or fasciculations and muscle wasting. Many of those who develop parkinsonism eventually evolve to the related syndromes of progressive supranuclear palsy (PSP) and corticobasal syndrome (CBS)37-43. A second major subgroup of about 10-15% of bvFTD patients develop motor neuron disease (MND), primarily with bulbar and proximal upper extremity impairment44-47.
On neuropsychological testing, bvFTD patients perform particularly poorly on tests of executive functions48-52. They are often impaired on digit span backwards, verbal fluency, Trail Making Test B, and the Tower Test. They do poorly on the Iowa Gambling Task, where they fail to improve when offered monetary rewards53. Patients with bvFTD cannot do tasks of abstraction and have poor verb comprehension relative to nouns54-56. They may be impaired on paradigms where they must infer other people’s mental states, thoughts and feelings, referred to as ‘theory of mind’57, and the ability to process social rule violations is impaired along with their sense of ‘personal’ morality30,58. In comparison, as previously noted, there is relative preservation of amnesia or visuo-spatial impairments on neuropsychological measures.
Progressive Nonfluent Aphasia
PNFA is characterized by an initial two or more year period of relatively isolated difficulty in verbal expression with agrammatism, difficulty with sentence syntax, shortened phrase length, and motor speech difficulties in the presence of relatively preserved comprehension (Table 57.2)59 – 62. Patients have difficulty with verbs and with sentence processing63. They also have apraxia of speech, with hesitant, broken and effortful output and phonologic (phonemic paraphasic) errors, particularly in repetition of polysyllabic words. Speech changes include progressive dysarthria with stuttering and oral apraxia64. Reading and writing is correspondingly impaired, and patients with PNFA are aware of their language and speech deficits.
Table 57.2 Progressive non-fluent aphasia
Source: Modified from Neary et al.61
| I. Core diagnostic features |
| A. Insidious onset and gradual progression |
| B. Agrammatism |
| 1. Grammatical morpheme and functor word omission |
| 2. Reduced mean length of utterance |
| 3. Phonemic paraphasias |
| C. Motor speech abnormalities |
| 1. Articulatory struggle and stuttering |
| 2. Dyarthria and/or dysprosody |
| 3. Apraxia of speech |
| II. Supportive diagnostic features |
| A. Impaired syntactical comprehension |
| B. Impaired repetition |
| C. Alexia and agraphia |
| D. Brain imaging: asymmetric abnormality predominantly affecting left hemisphere |
In PNFA, there is asymmetric atrophy of the left frontotemporal lobes61. Magnetic resonance imaging (MRI) with voxel-based morphometry (VBM) and functional imaging reveal marked atrophic changes or hypometabolism and decreased blood flow in the left peri- sylvian, inferior frontal and anterior insular areas59,65. Most patients with PNFA have frontotemporal neuropathology, but some patients have a language variant of AD at postmortem62,66.
Semantic Dementia
This FTD syndrome produces a multimodal loss of conceptual knowledge, particularly in word comprehension and facial recognition (Table 57.3). In contrast to PNFA, patients with SD have fluent speech and normal grammar but cannot understand words67. They have relative preservation of repetition, phonology, syntax, the ability to read aloud and the ability to write orthographically regular words. Their speech output becomes progressively empty, however, as they lose substantive words and their meanings. On testing, they have problems with single word comprehension on pointing commands, word definitions and matching of objects or pictures with words. On reading, there is a characteristic disorder termed ‘surface dyslexia’ with inability to read irregularly spelled words but preserved ability to sound them out68. The rest of their cognitive profile reflects semantic loss in other domains. There may be an impairment in the recognition of familiar faces (prosopagnosia) and impairment of object meaning or identity (object agnosia) that cannot be attributed to naming difficulties. Despite these deficits, SD patients can continue to learn (episodic memory) and have intact visuo-spatial skills69.
In SD there is circumscribed atrophy of the anterior inferior temporal gyri70. The cortical atrophy involves the left inferolateral temporal lobe and fusiform gyrus, right temporal pole, bilateral ventrome- dial frontal cortex, and the amygdaloid complex with sparing of the hippocampus70. Most SD patients have greater atrophy in the left anterior temporal region compared to the right. The asymmetric involvement is often evident on MRI imaging and correlates with relative impairments for word meaning (left temporal lobe) versus prosopagnosia or object recognition (right temporal lobe)71. The function of the anterior inferior temporal region appears to be to bind information together.
Table 57.3 Semantic dementia: consensus criteria
Source : Modified from Neary et al.61
| I. Core diagnostic features |
| A. Insidious onset and gradual progression |
| B. Poor confrontational naming, particularly for unfamiliar items |
| C. Loss of word comprehension, particularly for unfamiliar items |
| D. Intact word repetition |
| E. Intact motor speech AND/OR |
| A. Perceptual disorder characterized by |
| 1. Prosopagnosia: impaired recognition of identity of familiar faces |
| 2. Object agnosia: impaired recognition of object identity |
| B. Preserved perceptual matching and drawing reproduction Supportive diagnostic feature |
| Brain imaging: predominant anterior temporal |
| abnormality-bilaterally or asymmetric |
CLINICAL EVALUATION AND DIAGNOSIS
With the exception of the identifiable mutations, there are as yet no definitive tests for the FTD syndromes. In bvFTD, the behavioural changes outlined above usually precede or overshadow any cognitive deficits6‘18‘19‘72. Suspicion for bvFTD arises when there is a change in personality, usually in mid-life19‘73‘74. Suspicion for PNFA or SD arises when there is new speech or word-finding difficulty in mid-life. Clinical criteria for diagnosing the three main FTD syndromes are the FTD Consensus Criteria (see Tables 57.1-57.3)61. These criteria have proven imperfect’ with about 33-56% of bvFTD patients failing to meet Core Consensus Criteria on presentation (see Table 57.1)12‘13‘75. They are particularly deficient when it comes to a subgroup of bvFTD patients who lack changes on neuroimaging and who fail to progress after years of follow-up50‘51‘76. Currently’ the FTD Consensus Criteria are undergoing revision by an international team of experts in the field.
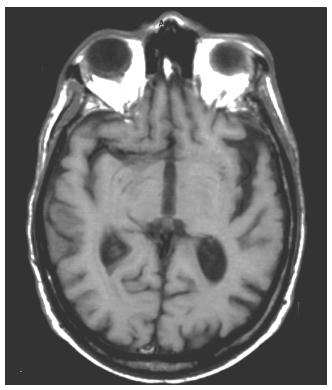
Brain MRI can help confirm the presence of an FTD syndrome (Figure 57.1)77. Although often normal in early stages78‘79’ MRI scans eventually show frontal and anterior temporal atrophy’ enlargement of the Sylvian fissures’ and anterior callosal atrophy80-86. There is temporal polar’ amygdalar and lateral temporal and fusiform gyral involvement but relative sparing of the hippocampi83‘87-90. FTD patients may have additional MRI evidence of bilateral caudate atrophy’ and others have changes in the substantia nigra and putamen or high signal changes in white matter91. VBM studies in autopsy- confirmed patients have confirmed the mesial frontal’ anterior insular and anterior temporal regional atrophy92-96. Finally’ recent studies suggest that the neuropathological variants of FTD may have different patterns of frontotemporal atrophy on neuroimaging95‘97.
Functional imaging may be more sensitive than MRI for the diagnosis of FTD. Positron emission tomography (PET) and single-photon emission tomography scans show decreased regional metabolism and cerebral blood flow in the frontal cortex and anterior temporal lobes (Figure 57.2)72‘82‘98‘99. On fluorodeoxyglucose (FDG) PET’ glucose uptake is reduced primarily in dorsolateral and ventrolateral prefrontal cortices and in frontopolar and anterior cingulate regions100. Although PET scan changes are not specific for FTD syndromes101’ worsening functional changes over time favour the diagnosis of a neurodegenerative dementia such as FTD102‘103. Furthermore’ new functional imaging technologies’ such as Pittsburgh Compound-B (PIB)’ hold great promise for distinguishing FTD syndromes from AD and other neurodegenerative disorders with amyloid-related pathology104‘105.
Figure 57.1 MRI (T2-weighted) image of progressive non-fluent aphasia (PNFA). Asymmetric atrophy of the left Sylvian fissure with reciprocal enlargement of the left frontal horn
There are important brain-behaviour relationships between symptoms and specific regions of involvement in FTD. Patients with bvFTD have greater right than left-sided atrophy106‘107. The initial pathology in bvFTD involves the ventromedial frontal cortex’ orbitofrontal region and anterior insula108‘109. Abnormalities in social and moral conduct correlate with changes in these regions on functional neuroimaging26. Apathy correlates with mesial frontal atrophy on both MRI VBM and on FDG-PET28110. Disinhibition correlates with atrophy of the subcallosal part of the frontal lobe’ orbitofrontal region and adjacent temporal areas28‘106‘110. PFNA and apraxia of speech correlate with left peri-Sylvian and anterior insular involvement92. SD correlates with anterior temporal atrophy88‘111. Moreover’ in SD’ impaired word comprehension corresponds to left anterior temporal atrophy’ and right temporal atrophy corresponds to impaired recognition of faces (Figure 57.3).
Most other biomarkers’ such as plasma TDP-43 or tau levels’ have not yet proven to be clinically useful for the diagnosis of FTD syndromes. Cerebrospinal (CSF) biomarkers hold the most promise for eventually helping distinguish FTD from AD112. Among patients with bvFTD, some have found a significant decrease of CSF tau protein113, but most report normal or only slightly increased CSF total tau and phospho-tau levels114-120. Overall, the literature indicates significantly lower total tau and tau/Aβ42 amyloid ratios in FTD compared to AD. In addition, decreased plasma levels of the PRGN protein in FTD patients with these mutations may be useful in screening for this specific genetic form of FTD (see below)121.
Figure 57.2 FDG-PET imaging of FTD patient. (a) Horizontal and (b) saggital views demonstrate frontal and anterior temporal hypometabolism
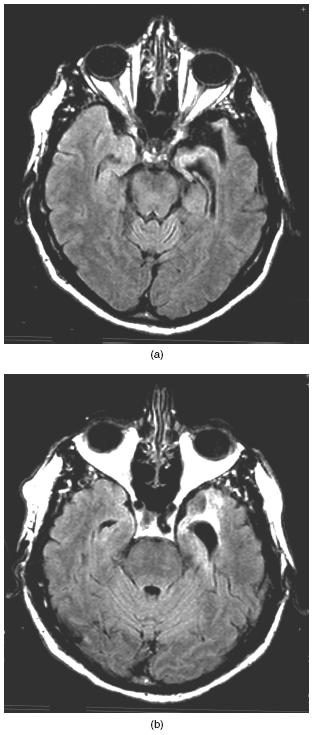
Figure 57.3 MRI (T2-weighted) images from patient with semantic dementia (SD). There is bilateral anterior temporal atrophy disproportionately affecting the left temporal lobe and associated with impaired word comprehension

There is frontotemporal lobar degeneration on gross pathology. The ventromedial frontal region and the anterior temporal areas have the most severe atrophic changes. Initially, the cortical degeneration is usually asymmetric and involves the mesial frontal regions including the anterior cingulate gyrus and the anterior insula. The frontotemporal cortex has neuronal and synaptic loss and astrogliosis with spongiosis (minute cavities or microvacuolation) of the outer, supra- granular (II-III) layers, with variable involvement of subcortical and limbic structures122-124. Specific loss of von Economo neurons occurs in the anterior cingulate and anterior insulae124. Finally, the serotoninergic and dopaminergic systems are decreased with relative sparing of cholinergic systems125,126.
The FTD syndromes include pathological variants that have intraneuronal inclusions that are either tau-positive or tau-negative, ubiquitin-positive (Table 57.4)127. FTD is most commonly due to tau protein, with tau-positive inclusions, or to the neuronal accumulation of TAR DNA-binding protein-43 (TDP-43) protein, with tau-negative, ubiquitin-positive inclusions. Among a cohort of 26 bvFTD patients, there were equal numbers with tau-positive and tau-negative pathology128, and among another 74 bvFTD patients, 55% were associated with a TDP-43 proteinopathy60,129. Tau is a microtubule-associated protein (MAPT) that stabilizes microtubules and promotes microtubule assembly by binding to tubulin130.
It is abnormal if hyperphosphorylated, resulting in a breakdown of microtubules and an impairment of axonal transport. TDP-43 is a nuclear protein whose function is not entirely clear, but appears to have a role in the regulation of transcription and mRNA splicing131. It is normal for TDP-43 to be ubiquinated, but it is abnormal when phosphorylated131.
Many patients with FTD syndromes have the intraneuronal accumulation of MAPT with tau-positive inclusions132-134. Six different isoforms of this tau protein result from alternative splicing, particularly of exon 10, which affects the number of carboxyl-terminal repeats135,136. An FTD ‘tauopathy’ may result from tau-positive inclusions due to either an imbalance in the ratio of tau isoforms with 3 or 4 microtubular binding repeats or to the abnormal hyperphosphorylation of tau. A tauopathy underlies nearly half of those with bvFTD, most of those with the PNFA variant, and all who develop parkinsonism, including the related disorders of PSP and CBS60‘92‘128‘129‘137.
More than half of patients with FTD syndromes have taunegative, ubiquitin-positive inclusions which contain TDP-43 (Figure 57.4)127,138-140
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
 Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


