INTRODUCTION
The alpha synucleinopathies encompass a diverse range of neurode-generative entities with the common neuropathological hallmark of abnormal accumulation in the brain of aggregates of insoluble alpha-synuclein. Major members of this club include Parkinson’s disease (PD), the associated Parkinson’s disease dementia (PDD), dementia with Lewy bodies (DLB), multisystem atrophy (MSA), pure autonomic failure and rapid eye movement (REM) sleep behaviour disorder. The clinico-pathological manifestation of each of these conditions appears to depend upon the pattern of insol-uble alpha-synuclein deposition in specific neuronal and glial populations1.
Of particular relevance to geriatric practitioners, and the focus of the present chapter, are the Lewy body dementias (LBDs) PDD and DLB, given that, combined, these diseases represent the second com-monest cause of dementia in the elderly2.
NEUROPATHOLOGYOFLEWYBODYDEMENTIAS
As intimated by their appellation, the common histopathological fea-ture of LBDs is Lewy bodies (LBs). These spherical neural inclusions were first described by Friederich Lewy in 1912 in the substantia nigra, locus coerulus and dorsal nucleus of vagus as well as in the hypothalamus, nucleus basalis of Meynert and sympathetic ganglia of PD patients3,4. However, interest in the role of LB formation in dementia did not arise until the latter half of the twentieth century when Japanese neuropathologists described a number of cases of dementia with severe extrapyramidal signs, who on autopsy had sig-nificant cortical LB deposition5–8. These cortical LBs can now be relatively easily visualized by the use of advanced neuropatholog-ical techniques including antiubiquitin and, more recently, specific alpha-synuclein immunohistochemical staining.
Histopathologically, LBs are eosinophilic neuronal inclusion bod-ies whose major constituent is aggregated alpha-synuclein bound together with ubiquitin and neurofilament. Cortical LBs tend to be smaller and less eosinophilic than LBs seen in the brainstem. It has been proposed by Braak and colleagues9 that in PD, LBs appear initially in the brainstem and then ascend progressively along the neuroaxis, with this pattern corresponding to the clinical develop-ment of parkinsonism. Later cognitive and neuropsychiatric symp-toms emerge then as a consequence of LB cortical involvement.
The temporal order of progression appears to be less clear in DLB although neuropathologically three categories, based on LB deposi-tion, have been proposed3: a brainstem-predominant DLB, a limbic (or transitional) DLB, and a predominantly cortical DLB. Correlation of these categories or the distribution of alpha-synuclein with demen-tia development, extrapyramidal signs or neuropsychiatric symptoms, however, is lacking10. In addition, current pathological criteria proba-bly do not explain the full range of potential pathological distributions that can occur, as up to 20% of older patients with LBs at autopsy have a cortical predominant pattern of distribution that does not con-form to either the Braak or DLB Consortium schemas11.
Neuropathological assessments of DLB are further complicated by the presence of co-existent Alzheimer’s disease (AD) pathology as high senile plaque counts are found in 80–90% of DLB cases; this is a comparable level to that found in pure AD. Conversely, LB pathology has been found in the amygdalae of individuals who have widespread AD pathology12 although these cases seldom display any clinical characteristics of DLB and thus should be regarded separately from LBDs3. These observations have, however, contributed to the sustained belief that AD and LBDs are caused by the same underlying disease as reflected in the use of terms such as ‘the Lewy body variant of AD’ for these mixed pathology cases13. This is inappropriate, since 80–90% of DLB cases neuropathologically do not have the typical AD-associated neocortical neurofibrillary tangles which are now known to be integral to the pathophysiology of AD14.More recently clinico-pathological data have suggested that the expression of a DLB clinical phenotype is related to the severity of LB pathology and inversely related to the severity of Alzheimer type pathology3,15.
CLASSIFICATIONOFLEWYBODYDEMENTIAS
Existing nosological systems such as DSM-IVR have been poorly operationalized with regard to PDD and DLB. PDD in DSM-IV-R is only briefly referenced and its syndromal definition is imprecise requiring the presence of PD with associated cognitive and motoric deficits, executive dysfunction and impairment in memory retrieval, which may be exacerbated by depression. DLB as a disease entity is not even listed in DSM-IV-R and this probably reflects the presumed association with AD as well as the various and confusing diagnostic labels which have been attached to this condition over the years, including Lewy body dementia, Lewy body variant of AD, senile dementia of Lewy body type and diffuse cortical Lewy body disease.
An important step forward to address these issues has been the for-mulation of operationalized criteria for both PDD16 and DLB5,6 (see Tables 58.1, 58.2 and 58.3), with the latter undergoing a revision in 20053. These have allowed clinicians to make more precise diagnoses and develop clearer management strategies. From the perspective of research, operational criteria have begun to facilitate in a system-ized manner a better understanding of the natural history of these conditions and improved clinico-pathological correlations, as well as allowing clinical trials to be conducted to standards accepted by drug regulatory authorities. Overall, however, it is important to rec-ognize that the criteria for DLB and PDD are still under development and will require further refinement (e.g. neuropathological criteria), as well as being complemented by effective disease biomarkers to improve early detection (see later discussion).
As presented in Tables 58.1 and 58.2, the diagnosis of probable or possible PDD depends primarily upon the presence of core symptoms of (i) diagnosis of parkinsonism, (ii) a dementia with an insidious onset and slow progression in the context of established PD and (iii) the absence of features which could suggest other conditions as the cause of mental impairment16. The diagnosis of probable PDD depends upon the presence of the core symptoms as well as a cogni-tive profile typical for PDD (e.g. attention, visuo-spatial) and at least one behavioural symptom (e.g. visual hallucinations). The diagno-sis of possible PDD is less rigorous; while still requiring the core features, the associated cognitive impairment may be, for example, atypical; behavioural symptoms may or may not be present and there may be features which make the diagnosis less robust, e.g. evidence for vascular disease on imaging. For a diagnosis of probable or possible DLB (Table 58.3), consensus criteria demand that the core features of fluctuating cognitive impairment, recurrent visual hal-lucinations and parkinsonism should be present (two or more core features for a diagnosis of probable DLB and one core feature for a diagnosis of possible DLB). DLB criteria also provide supportive and suggestive features that may make the diagnosis more likely. Clinical assessment of these features is discussed in more detail later.
Table 58.1 Features of dementia associated with Parkinson’s disease
Source: From Emre etal. (2007); Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement Disorders 22(12):1689–1707. Reprinted with permission of John Wiley & Sons, Inc.

Table 58.2 Criteria for the diagnosis of probable and possible PD-D
Source :From Emre etal. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. MovementDisorders22(12): 1689–1707. Reprinted with permission of John Wiley & Sons, Inc.

Table 58.3 Consensus criteria for the clinical diagnosis of probable and possible DLB
Source : Adapted from McKeith etal. Diagnosis and management of dementia with Lewy bodies. Third report of the DLB consortium, Neurology2005; 65: 1863–72. Reprinted with permission from Wolters Kluwer Health.

The development of these criteria has, however, not been without difficulties:
Deciding if it is DLB or PDD
The diagnostic separation of DLB from PDD, on the basis of when the motor features occur relative to the dementia has caused controversy17–20 . Currently both the consensus criteria for DLB and PDD recommend that for a diagnosis of PDD, the extrapyra-midal motor features need to be present for at least 12 months or more before the onset of the dementia, but if the dementia precedes the motor symptoms or occurred within 12 months of the motor features then the diagnosis should be DLB. This boundary delineation has been considered useful in research for determining the epidemiology and natural history of these conditions as well as providing clinicians with clear categorical labels for diagnosis. However, separation of PDD and DLB on a temporal arbitrariness has no strong clinical or pathological basis. Indeed, as will be discussed below, while minor differences do exist between DLB and PDD, they demonstrate remarkably similar cognitive profiles as well as similar neuropsychiatric features, neuroleptic sensitivities, sleep symptoms, autonomic dysfunction and responsiveness to cholinesterase inhibitors21. Overall unitary approaches have proven useful for study of common neurobiological and genetic processes in these conditions, and, indeed, the term Lewy body disease, which encompasses PD, PDD and DLB, and the term Lewy body dementias, which includes PDD and DLB, have been particularly helpful in this regard (see Figure 58.1). Therefore, while separating the dementias, PDD from DLB, avoids confusion clinically and helps stream patients depending upon the clinical setting in which they are first diagnosed (e.g. movement disorder clinics for PDD and dementia clinics for DLB), in certain cases, particularly when the temporal ordering of motor to cognitive symptoms is not clear, the umbrella term LBDs is likely to be more useful (Table 58.4).
Problems with Validation of DLB Consensus Criteria
Neuropathological validation studies using the 1996 DLB consensus criteria6 suggested that while the specificity of the clinical diagnosis of probable DLB, i.e. LB pathology found at autopsy, is high at >80%, the sensitivity ?60%, which suggests that many LB pathol-ogy cases do not present with the typical features of fluctuations, visual hallucinations or parkinsonism. Difficulties in clinically dis-cerning core symptoms in certain individuals, particularly when the dementia is severe, the fact that significant LB pathology may occur later in the disease course such that its clinical effects are limited, or the co-occurrence of Alzheimer pathology, which modifies the clinical presentation, are all possible explanations for this reduced sensitivity in DLB detection. Importantly, it means that a significant proportion of potential DLB cases are missed; however, biomarkers and, in particular imaging techniques (see below), which enhance sensitivity in atypical/uncertain cases, may help overcome this problem.
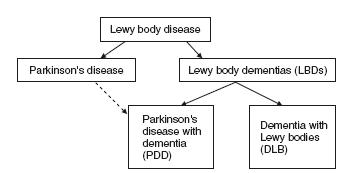
Table 58.4 Key points in classification
| • Parkinson’s disease with dementia (PDD) should be used todescribe dementia that occurs in well-defined Parkinson’s |
| • Dementia with Lewy bodies (DLB) should be diagnosed when the dementia occurs before or concurrently with parkinsonism |
| • In research, the parkinsonism needs to be present for at least one year before the onset of dementia to make a diagnosis of PDD, but if the dementia occurs within 12 months of the motor symptoms, or indeed precedes it, then the diagnosis should be DLB |
| • Lewy body disease includes PD, PDD and DLB |
| • Lewy body dementias (LBDs) are part of the Lewy bodydisease spectrum and include PDD and DLB |
| • When it is difficult to diagnostically separate PDD from DLB,the generic term LBDs might be more helpful clinically. |
The aetiology of LBDs remains unclear. However, a number of biological candidates have been proffered, including LB formation, alpha-synuclein pathology and neurochemical deficits.
LB and Alpha-Synuclein Pathology
Initially LBs were hypothesized as the core neurotoxic lesion involved in the aetiology of LBDs given their association in PD with nigro-striatal dopaminergic cell loss. However LBs are only sparsely distributed in the cortex in LBDs and their density does not appear to correlate strongly with disease duration or symptom22.More recent thinking has been to view LBs as a neuroprotective response by vulnerable neurons attempting to sequester toxic alpha-synuclein species. An earlier stage in the neurodegenerative process appears to be the accumulation of small aggregates of alpha-synuclein. These aggregates, which are enormous in number, appear to be more widely distributed in the brains of patients with PDD and DLB than LBs and their presence in presynaptic terminals is thought to severely impair synaptic function23. The presence of amyloid beta in the neocortex has also been associated with more extensive alpha-synuclein lesions and thus it has been speculated that amyloid beta may have a role in the genesis of alpha-synuclein deposition in LBDs. This is an intriguing finding as it may provide evidence of a link between LBDs and AD pathology but further work is required to clarify this interaction24.
Recently, a great deal of interest has been focused on the role of the enzyme glucocebebrosidase (GBA) in LBDs. GBA deficiency as a result of an autosomal recessive mutation has long been known to be associated with Gaucher’s disease, where there is marked cellular accumulation of the GBA lipid substrate, glucocerebroside. How-ever, it has become evident that while the severe manifestations of Gaucher’s occur when there is less than 20% GBD activity, milder heterozygous mutant variants can have little deleterious effect in early life but can cause the later development of PD or DLB (see below for further discussion)25. Currently it is unclear in what way GBA mutations are related to PD and LBDs although it has been specu-lated that impaired ceramide metabolism (in which GBA is involved) and lysosomal recycling interact with the process of alpha-synuclein formation.
Neurochemical Changes
In addition to the neuropathological changes, there are severe deficits in monoaminergic and cholinergic systems in LBDs (see O’Brien etal. for further elaboration2). While the link between nigro-striatal dopaminergic neuronal degeneration and parkinsonism is established, it is also speculated that abnormal dopamine transmission, particu-larly in the striatum and frontal areas, contributes to the executive dysfunction26. Cell loss in raphe nuclei (serotonin) and locus coerulus (norepinephrine) may explain the mood symptoms, apathy and atten-tional dysfunction evident in LBDs. Cholinergic deficits in LBDs have been suggested to contribute to the global cognitive impair-ment and attentional dysfunction as well as hallucinosis; amelioration of these symptoms by cholinesterase inhibitors (ChEis) strongly sup-ports the involvement of cholinergic dysfunction in LBDs pathology.
Parkinson’s Disease Dementia
The prevalence of dementia in PD has been estimated at between 24% and 31%, and the incidence rate is at least four to six times that of the rate of dementia in age-matched controls27,28. It is becoming increasingly recognized that cognitive impairment is present even in the earliest stages of PD; in two recent studies it was estimated that between 19% and 24% of newly diagnosed PD patients appear to have some degree of cognitive impairment29,30. The nature of these deficits varies, but subtle deficits in executive function, including attention shifting and working memory, as well as deficits in visuo-spatial function and episodic memory, have been noted. With time there appears to be a broadening and exacerbation of these deficits and it has been suggested that the mean duration from onset of PD to the development of PDD is around ten years; indeed one longitu-dinal study suggested that the cumulative prevalence was up to 78% after eight years of follow-up31. Risk factors for the development of PDD include older age, mild cognitive and attentional impairments at the initial diagnosis of PD, the severity of motor manifestations, and, in particular, the presence of rigidity, postural instability and gait disturbance32–35 . While PD incidence is twice as common in men as women it is not clear if male gender predisposes PD patients to the development of PDD. Smoking may also be a risk factor but the presence of apolipoprotein E4 (APOE4) genotype, unlike AD, does not appear to be a risk factor for PDD36–39. Recent large-scale autopsy studies have demonstrated that between 4% and 10% of PD patients have evidence for GBA mutations. These individuals tend to display early-onset parkinsonism and at least half had halluci-nations and cognitive decline; it has recently been suggested that GBA mutations represent the most common genetic risk factor for the development of PD or LBDs40.
Dementia with Lewy Bodies
Community prevalence figures for DLB have ranged from 0% to 30.5% of dementia cases depending on the sample cohort41.This wide range can be explained by a small number of studies with low patient numbers, differences in case criteria used and the pres-ence of significant sampling biases. However, studies have suggested approximately 15–30% of all dementia cases meet neuropathological criteria for DLB at autopsy, emphasizing that a significant proportion of DLB cases are probably being missed clinically42. There are very few studies examining the incidence of DLB; two longitudinal stud-ies, one US-based and the other Japanese-based, estimated the inci-dence as 0.57 per 1 000 person years and 1.4 per 1 000 person years, respectively43,44. Risk factors for DLB are unclear as prevalence stud-ies have been too small. It has, however, been suggested that APOE4status reduces survival rate in DLB (as with AD) although its pres-ence has little effect on disease onset or duration45. With regard to GBA, an autopsy study that included 95 patients with pathologi-cal confirmed DLB noted that 28% of subjects had GBA mutations. Those patients with AD co-pathology had substantially less at 10%, suggesting that GBA mutations tend to lead to more pathologically pure forms of LBDs46. There may be an increased prevalence of DLB with older age and some case series have suggested a male preponderance although more recent studies have refuted this. Of note, however, is a recent neuropathological study suggesting that while the prevalence of LBs is almost the same between the sexes, the severity of LB pathology may differ, with males having a more brainstem/limbic pattern compared to a more diffuse neocortical pat-tern seen in women15.
LBDs have a distinctive constellation of symptoms and signs, including progressive cognitive impairment, neuropsychiatric alter-ations, parkinsonism, sleep behaviour disturbances and autonomic dysfunction. In PDD, parkinsonism is the initial complaint with the subsequent development over many years of an insidiously progressive cognitive impairment. Neuropsychiatric symptoms such as visual hallucinations are often initially viewed as a side effect of dopamine replacement therapy, but later, particularly as the cognitive impairment advances, they often arise independent of any anti-parkinson treatment. DLB patients, on the other hand, often have less prominent parkinsonism at initial presentation; instead, they tend to present with an evolving dementia that is often associated with attentional/cognitive fluctuations as well as visual hallucinations. These differences in the clinical presentation of the PDD versus DLB, particularly earlier in the disease course, are likely to relate to the differences in pathological spread and distribution of LBs and alpha-synuclein between the two conditions, as previously discussed. However, symptom and feature convergence occur between PDD and DLB later in the disease course, reflecting the commonality at this point in terms of neuropathology.
Cognitive and Attentional Impairment
Clinicians with experience in LBDs often state that they get an inherent ‘feel’ of how the cognitive profile of patients with LBDs differs from that of patients with AD. Patients with LBDs have been suggested to display ‘subcortical’ features such as bradyphrenia and attentional deficits as well as visuo-perceptual problems (dispropor-tionate to the global cognitive impairment), and this contrasts with the more ‘cortical’ feel of AD, where language and memory deficits predominant. Thus on the Mini-Mental State Examination, a patient with DLB or PDD might be well orientated to time and place, score two out of three points on the delayed recall, but be unable to perform the serial sevens or copy the double pentagons.
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
 Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


