Organ involved
Specific tumor (prevalence by 40 years)
Clinical presentation
MEN1
Parathyroid disease (95 %)
Diffuse hyperplasia
Symptoms related to hypercalcemia or hypercalciuria
Adenoma, multiple
NETs
PNETs (30–80 %)
Gastrinomas (>50 %)
ZES, diarrhea, abdominal pain
Insulinomas (10–30 %)
Whipple triad
Glucagonomas (~3 %)
Necrolytic migratory erythema, weight loss, anemia, stomatitis
VIPomas (extremely rare)
Verner-Morrison syndrome
NF PNETs (20–100 %)
Asymptomatic, but with malignant potential
Somatostatinomas (extremely rare)
Somatostatinomas syndrome, rare
Other (e.g., GHRH-secreting)
Rare, increased GH and IGF1 levels
Foregut NETs (2–10 %)
Thymic, gastric, bronchial NETs
Organ specific
Pituitary tumors
Prolactinomas (20 %)
Oligomenorrhea, galactorrhea, infertility in woman; impotence and infertility in men
Other: ACTH, TSH, GH+PRL, GH, NF (each 2–9 %)
Hormone-dependent
Other endocrine manifestations
Benign adrenocortical tumors (73 %)
Most nonfunctioning
Adrenocortical carcinoma (13 %)
Hormone-dependent
Pheochromocytomas (<1 %)
Rarely described, mainly asymptomatic
Thyroid adenomas, goiter, and carcinoma (25 %)
Usually incidental finding
CNS tumors
Ependymomas, schwannomas, meningiomas (1 %)
Mainly asymptomatic
Cutaneous manifestations
Multiple subcutaneous lipomas (33 %); visceral, pleural, or retroperitoneal lipomas (rare)
Facial angiofibromas and collagenomas (up to 88 %)
20.2.1.1 Clinical Features and Diagnosis
PHPT is the most common and frequently the earliest manifestation in MEN1 mutation carriers and occurs in ∼95 % of cases [9, 13]. The PHPT in MEN1, as compared to the sporadic form, has an earlier age of onset (20–25 years, compared to 55–60 years), exhibits an equal male to female ratio (compared to a female preponderance in sporadic PHPT), and involves all four parathyroid glands (diffuse hyperplasia or multiple adenomas), rather than a single one (solitary adenomas) [8, 9]. Noteworthy is the increased frequency of supernumerary (up to 20 %) and ectopic parathyroid glands in MEN1 patients, usually localized within the thyroid gland, in the anterior mediastinum, or exceptionally in the pericardium [18]. Although the MEN1 gene is a tumor suppressor, parathyroid carcinoma may be diagnosed in a small percentage of patients [19].
PHPT is a progressive disease in MEN1: whereas asymptomatic hypercalcemia is the most common manifestation, patients may present with symptoms (e.g., fatigue, weakness, polydipsia, polyuria, myalgias, or abdominal pain) or signs (nephrolithiasis or osteitis fibrosa cystica) related to the progressive increase in serum and/or urinary calcium and disease duration.
A major clinical manifestation of MEN1-associated PHPT is progressing demineralization with severe osteopenia (T score < –2.0) in ~ 44 % of patients with uncontrolled disease, by the age of 35 [20]. Recurrent kidney stones are less frequently described in MEN1 families, and it is unusual for these patients to develop chronic renal failure as a result of nephrolithiasis [21, 22].
The diagnosis of PHPT is based upon demonstrating elevated serum levels of calcium and parathyroid hormone (PTH). Mild hypercalcemia with normal/increased range of serum PTH concentration can usually be detected during the second decade of life [23]. Total serum calcium concentration corrected for albumin level is considered the screening test for PHPT in MEN1 [24]. Noteworthy, identifying the “subclinical” stage of hyperparathyroidism has not been considered essential until recently, when an increased cardiovascular risk has been reported in patients with mild normocalcemic hyperparathyroidism [25]. However, the performance of serum PTH measurement as a screening test remains controversial. Diagnosis of hyperparathyroidism requires levels of PTH inadequately high for the concomitant calcium levels. At present, different kits for assessment of PTH are available. Second generation of kits, measuring concentrations of the so-called intact PTH (iPTH), is in general usage [26]. Routine testing for MEN1 mutations in young patients with primary hyperparathyroidism is not recommended, as these mutations are rare in unselected patients even below 40 years of age [27]. Such testing should be considered in patients with multiple gland involvement, coexistence of other tumors characteristic for MEN1, or family history of hyperparathyroidism or MEN1 tumors.
20.2.1.2 Treatment of MEN1-Associated PHPT
In contrast to single-gland resection in sporadic cases, a more complex surgical approach, including total parathyroidectomy and thymectomy with autotransplantation of parathyroid tissue or excision of 3.5 parathyroid glands, is indicated for PHPT in MEN1 [18, 28, 29]. Moreover, postoperative hypoparathyroidism and higher rates of recurrent or persistent disease are more frequent in MEN1-associated PHPT than in the sporadic form of the disease [28]. Recurrence is usually located in preserved/residual parathyroid tissue [30], and it is strongly influenced by the surgeon’s experience and the possibility to perform intraoperative PTH determinations [28]. Intraoperative PTH assessment is mandatory in these patients to confirm the removal of all functional parathyroid tissue before parathyroid autotransplantation.
In MEN1 patients with preclinical/eucalcemic PHPT, the approach is still uncertain: early parathyroidectomy is recommended by some experts to control even mild hyperparathyroidism and to prevent the progressive decline of bone mineral density and the increased risk for kidney stones [31, 32].
In MEN1-associated PHPT patients in whom surgery is not possible or ineffective, medical treatment with calcimimetic drugs emerged as an effective, though expensive, option [33, 34]. Cinacalcet therapy is well tolerated by most of the patients; rarely, gastrointestinal side effects may induce discontinuation of the therapy [34, 35]. Interestingly, the administration of the somatostatin analogue octreotide LAR controlled hypercalcemia and hypercalciuria in two-thirds of patients with MEN1-related PHP, this effect being most probably mediated by the octreotide LAR binding to somatostatin receptors on parathyroid tumor cells in patients with MEN1-related PHP [36].
20.2.2 MEN1-Associated Pancreatic Islet Cell Neuroendocrine Tumors (PNETs)
Pancreatic and duodenal neuroendocrine tumors represent the second most frequent manifestation in MEN1 (occurring in 30–80 % of cases) and continue to be the primary cause of tumor-related death in these patients [10, 37]. Although PHPT is usually the first manifestation of MEN1 syndrome, the penetrance of MEN1-related PNETs is similar to that of parathyroid tumors [38]. In contrast with the sporadic PNETs, MEN1-related PNETs are characterized by an early onset, multifocality, variable expression of the tumors, and propensity for malignant transformation; both the histology and the size of the tumor are predictors of the malignant potential [5, 39–41]. Some of these tumors secrete gastrin, insulin, glucagon, vasoactive intestinal polypeptide (VIP), etc., and are associated with distinct clinical syndromes, whereas most PNETs are clinically silent tumors (nonfunctioning, secreting pancreatic polypeptide, PP) [5]. Given that MEN1-associated PNETs are usually multiple and of a possible malignant behavior, their timely diagnosis and management are challenging and of major importance.
20.2.2.1 Gastrinomas
Clinical Manifestations and Diagnosis
Gastrin-secreting tumors (gastrinomas) are the most frequent functioning duodenopancreatic NETs in patients with MEN1 (>50 %), and ~ 20 % of patients with gastrinomas will have MEN1 [42–44]. Gastrinomas occur more often in patients with MEN1 who are older than 30 years; are associated with marked gastric acid secretion and recurrent and severe peptic ulcerations (the Zollinger-Ellison syndrome, ZES), which may perforate; and are usually metastatic at diagnosis [45, 46]. These features are responsible for the major morbidity (peptic ulcer disease, diarrhea and steatorrhea, cachexia, and abdominal pain) and mortality associated with MEN1 [47, 48].
In contrast to the sporadic gastrinomas that occur predominantly in the pancreas, in MEN1, most of these tumors develop in the proximal duodenum [49]. Gastrinomas are usually small (<5 mm) and multiple, growing slowly deep into the mucosa and metastasizing frequently to the peripancreatic lymph nodes and further to the liver [50]. Pancreatic gastrinomas or other extra-pancreatic, extra-duodenal (e.g., ovary, liver, bile duct, pylorus, spleen, mesentery), and extra-abdominal (e.g., cardiac intraventricular septum) locations in MEN1 patients are extremely rare [51, 52]. When found in the pancreas, it may be difficult to distinguish gastrinomas from concomitant nonfunctioning PNETs, and in this eventuality selective arterial secretagogue injection test (SASI test) may help in localizing the gastrin-secreting tumor [43, 53].
The diagnosis is established by demonstration of an increased fasting serum gastrin concentration (found to be elevated in 90–98 % of ZES patients) after stopping antisecretory drugs (proton pump inhibitors, PPIs, at least 1 week; histamine receptors 2 blockers, H2R, for at least 2 days) [54], in association with increased basal gastric acid secretion (gastric pH < 2) [52, 55]. However, stopping antisecretory drugs in ZES patients is often impossible due to severe symptoms of peptic disease and diarrhea and may be hazardous due to possible perforation or bleeding. Occasionally, iv provocative tests with either secretin (2 U/kg) or calcium infusion (4 mg Ca2+/kg∙h for 3 h) are required to distinguish patients with ZES from other patients with hypergastrinemia (e.g., those with antral G-cell hyperplasia) [50]. In two-thirds of patients with ZES, fasting serum gastrin levels overlap with values seen in other conditions. In these patients, gastrin provocative tests are needed to establish the diagnosis of ZES [56]. For unknown reasons, secretin stimulates the release of gastrin by gastrinoma cells, and therefore patients with these tumors have a dramatic rise in serum gastrin in response to a secretin infusion. In contrast, normal gastric G cells are inhibited by secretin, and therefore serum gastrin concentrations do not rise in patients with other causes of hypergastrinemia. The secretin stimulation test is performed by administering 0.4 μg of secretin per kg body weight (RepliGen Corporation, Waltham, MA) intravenously over 1 min; a baseline serum gastrin is measured twice before the secretin is administered and classically 2, 5, 10, 15, and 20 min later. An increase of ≥120 pg/mL in gastrin levels is highly suggestive of gastrinoma [56].
In some patients with MEN1, the diagnosis of ZES may be difficult, as it does not appear to develop in the absence of primary hyperparathyroidism, whereas hypergastrinemia has also been reported to be associated with hypercalcemia [54]. Moreover, successful treatment of MEN1-associated PHPT with restoration of normocalcemia is known to ameliorate clinical symptoms and biochemical abnormalities in ~ 20 % of MEN1 patients with ZES [44].
Tumor localization and determination of the tumor extent are essential to the proper management for MEN1 gastrinomas: endoscopic ultrasonography, CT, MRI, selective abdominal angiography, and mainly somatostatin-receptor scintigraphy/Ga68-DOTA-TATE/-TOC/-NOC PET-CT are very helpful in localizing the tumor [53, 57]. Moreover, the combination of intra-arterial calcium injections with hepatic venous gastrin sampling has been shown to regionalize the gastrinomas [58]. Importantly, these tumors are often occult to conventional exploration, and their detection requires duodenotomy and meticulous evaluation of the mucosa by eversion and direct palpation [59].
Management of MEN1-Associated Gastrinomas
Control of Gastric Hypersecretion
The management of patients with ZES is directed toward reducing basal acid output to less than 10 mmol/l and includes acute- and long-term control of gastric acid hypersecretion by parietal cell H+-K+-adenosine triphosphatase (proton pump) inhibitors, PPIs (omeprazole, lansoprazole, pantoprazole, rabeprazole, and esomeprazole), starting a dose equivalent to 60 mg/day of omeprazole [54]; the dosage should be increased to b.i.d. in patients with severe gastroesophageal reflux disease (GERD) symptoms, with previous Billroth II or gastroenterostomies, or with hypercalcemia with MEN1 [52]. Long-term PPI treatment is safe and without loss of efficacy; no gastric carcinoid development associated to PPI-induced hypergastrinemia was demonstrated in humans [52]. Some patients may also require additional treatment with the histamine H2 receptor antagonists, cimetidine or ranitidine [5].
Surgical Management of Gastrinoma
In patients with MEN1 and ZES, correction of the PHPT by parathyroidectomy can significantly decrease basal acid output and fasting gastrin level and increase sensitivity to gastric antisecretory drugs [60]. The role of surgery in the treatment of the gastrinoma in MEN1/ZES patients is controversial [61, 62]: these patients cannot be cured as they usually have multiple duodenal tumors with frequent lymph node metastases. In addition, they usually have concomitant nonfunctional PNETs. As development of liver metastases, associated with a poor prognosis, is more frequent in the presence of large tumors, exploration is recommended whenever a PNET of >2 cm is found [62]. Surgical resection of duodenal MEN1-associated gastrinomas, based on preoperative and intraoperative (e.g., transillumination of the duodenum) tumor regionalization, may be considered in specialized centers. Due to increased morbidity and potential mortality after extensive resections, the surgical excision has to be individualized based on preoperative findings, patient history (e.g., preexisting diabetes), and preference [37]. Interestingly, the presence of lymph node metastases in 50–60 % of patients with MEN1-related duodenal gastrinomas is not usually associated with a decreased survival [60].
Management of Metastatic MEN1 Gastrinoma
Long-acting somatostatin analogues (SSAs) such as octreotide LAR, lanreotide SR, or the lanreotide Autogel alone or in combination with interferon are recommended as initial therapy [52, 63]. The main effect of these agents is tumoristatic effect, with disease stabilization observed in ~ 40–70 % of patients, whereas decrease in tumor size (partial response) is reported in only 10–20 % of patients [64]; these agents are particularly effective in slower-growing PNETs [65]. Chemotherapy with streptozotocin and 5-fluorouracil, hepatic artery embolization, PRRT, or removal of all resectable tumor have been occasionally successful in patients with disseminated MEN1 gastrinomas [66, 67].
20.2.2.2 Insulinomas
Clinical Manifestations and Diagnosis
Insulinomas represent between 10 and 30 % of all MEN1-related PNETs, and rarely (in 10 % of patients with MEN1), they may coexist with a gastrinoma or with other nonfunctioning PNETs at the time of diagnosis, in the same patient [9, 68]. In MEN1 patients, insulinomas occur more often at an age younger than 40 years (in many patients even younger than 20 years and in some as young as 5 years), in contrast to the sporadic insulinoma patients in whom the disease occurs usually after the age of 40 years [9, 69]. Insulinoma is the first manifestation of MEN1 in 10 % of patients, whereas ~ 4 % of patients with insulinomas will have MEN1 [5, 66]. The clinical presentation, biochemical and anatomical diagnosis of MEN1-related insulinomas, and the medical and surgical treatment of these patients are similar to those in the sporadic insulinoma patients and are addressed in detail in the “Insulinomas” chapter of this textbook [70, 71].
20.2.2.3 Glucagonomas
Clinical Manifestations and Diagnosis
Glucagon-secreting pancreatic tumors occur in fewer than 3 % of patients with MEN1 (Table 20.1) [5, 50], are highly malignant, and frequently are localized at the tail of the pancreas [5, 72]. Importantly, in MEN1-associated glucagonomas, the characteristic clinical manifestations (such as the necrolytic migratory erythema, weight loss, anemia, or stomatitis) [73, 74] may be absent, and the tumor may be detected in a completely asymptomatic patient with MEN1 undergoing routine pancreatic imaging or detected by glucose intolerance and concomitant increase in glucagon levels [75, 76].
Management of MEN1-Associated Glucagonomas
Surgical removal is the treatment of choice. However, up to 80 % of glucagonoma patients have metastatic disease at the time of diagnosis [77], and therefore systemic therapy using SSAs, PRRT, chemotherapy (streptozotocin and 5-fluorouracil), or hepatic artery embolization has been successful in some patients [63, 78–81].
20.2.2.4 VIPomas
Clinical Manifestations and Diagnosis
The vasoactive intestinal peptide (VIP) secreting tumors (VIPomas) are rare in MEN1, occur commonly in the tail of the pancreas, and produce a clinical syndrome (Verner-Morrison syndrome) with watery diarrhea, hypokalemia, and achlorhydria (WDHA) [80, 82]. The diagnosis requires excluding laxative and diuretic abuse, demonstration of a large fasting stool volume (0.5–1 l/day) during a fast, and an elevated serum VIP concentration [50].
Management of MEN1-Associated VIPomas
Surgical excision of VIPomas is the main therapeutic intervention in these patients, with a curative purpose. However, in patients with unresectable or metastatic tumor, systemic treatment with SSAs, chemotherapy (streptozotocin and 5-fluorouracil), corticosteroids, indomethacin, metoclopramide, lithium carbonate, and hepatic artery embolization has been used with different efficacy [37, 38].
20.2.2.5 Nonfunctioning PNETs (NF PNETS)
Clinical Manifestations and Diagnosis
NF PNETs represent a heterogeneous group of tumors being increasingly identified as the result of increasing sensitivity of radiological screening methods [40, 41, 83]. Importantly, NF PNETs have been reported to occur in young asymptomatic patients who are even less than 15 years of age. NF PNETs are not associated with a clinical syndrome and may be associated with minor elevation of specific pancreatic hormones (e.g., pancreatic polypeptide) but without clinical symptoms [38].
Identification of NF PNETs is of outmost importance, as they are the most common NETs occurring in the MEN1 setting, they have a well-recognized malignant potential, they are associated with a worse prognosis, and they are the most common cause of death in MEN1 patients [47, 55, 84, 85]. In the absence of specific clinical and biochemical abnormalities, the diagnosis of NF PNETs may be delayed, and therefore radiological screening for enteropancreatic NET in MEN1 is mandatory and should begin at the age of 10 years. The optimal screening method and its timing interval are still controversial and depend on method availability and skills of the performer [48, 83]: endoscopic ultrasound is probably the most sensitive, whereas somatostatin-receptor scintigraphy/gallium-68-DOTA-TATE/-TOC/-NOC PET-CT is the most specific method for detecting metastatic disease. The clinical significance of small NF PNETs (e.g., <1 cm) in asymptomatic individuals is still not fully understood [86].
Management of MEN1-Associated NF PNETs
Surgical Therapy of MEN1-NF PNETs
The treatment approach of asymptomatic NF PNETs is controversial. To date, treatment decision is based on tumor size, as the risk of metastases is thought to be higher in patients with larger tumors [87, 88]. Some reports suggest that early and aggressive surgery might reduce the risks for malignant progression [89]. However, other studies have not confirmed this association [45, 90], and therefore, there is no consensus to date regarding the indications and timing of surgery. The latest clinical guidelines [37] suggest considering surgical resection for NF PNETs that are more than 1 cm in size which may be successful in up to 80 % of MEN1 patients [91, 92]; however, other centers recommend surgery only for tumors of more than 2 cm [87]. For tumor size less than 1 cm, surgical resection should be considered for tumors with a size doubling time of a 3- to 6-month interval [37]. However, there are many factors influencing the decision on surgery in these patients, such as the increased morbidity of pancreatic surgery (as complications such as diabetes mellitus, steatorrhea, dumping syndromes are not uncommon) [38], the possible presence of occult metastatic disease at the time of initial diagnosis, and the high risk for disease recurrence in the remnant pancreatic tissue after surgery [91–93].
Medical Therapy of MEN1-NF PNETs
PNET cells may express different kinase receptors, such as tyrosine kinase receptors (TK-R), vascular endothelial growth factor receptors (VEGF-R), and platelet-derived growth factor receptors (PDGF-R), whereas some exhibit over-activation of the mammalian target of rapamycin (mTOR) signaling pathway, which stimulates cell proliferation and angiogenesis. Recently, systemic therapy with tyrosine kinase receptor inhibitors (TKI; sunitinib malate) or with mTOR inhibitors (mTORi; everolimus) was reported as significantly increasing time to tumor progression in patients with locally progressive or metastatic PNETs [94, 95]. Moreover, all the other treatment modalities considered for functioning PNETs such as chemotherapy (the classic streptozotocin and 5-FU combination or the new combination of temozolomide and capecitabine), SSAs, peptide receptor radioligand therapy, hepatic artery chemoembolization, radio-frequency ablation, etc., may be individually used in treating patients with metastatic NF PNETs [96–100].
20.2.2.6 Other PNETs
Rare PNETs ectopically secreting growth hormone-releasing hormone (GHRH) have been reported in some patients with MEN1 [66, 101]. They may occur in the lungs, pancreas, or the small intestine and should be suspected when elevated levels of growth hormone (GH) and GHRH are found in the serum of the patient or when the patient has clinical acromegaly.
About 0.65 % of MEN1-related PNET may produce somatostatin and are defined as somatostatinomas, rare tumors which are usually nonfunctioning or, in 10 % of cases, associated with a clinical syndrome of hyperglycemia, cholelithiasis, low acid output, steatorrhea, diarrhea, abdominal pain, anemia, and weight loss (the somatostatinoma syndrome) [72, 102].
Surgical removal is the treatment of choice for these rare tumors.
20.2.3 MEN1-Associated Pituitary Tumors
Patients affected by the MEN1 syndrome display a high incidence (~42 %) of pituitary adenomas [103–105], which may occur as early as 5 years of age and as late as the ninth decade and have been reported to occur more frequently in women than men (Table 20.1) [103]. The molecular pathways involved in their development seem different from the sporadic tumors [106–108]; approximately 40 % of patients with MEN1 have pituitary adenomas and 17 % present with a pituitary tumor [109].
All types of adenomas can be found in MEN1 patients with a predominance of prolactinoma and macroadenoma (i.e., diameter greater than 1 cm) compared to a control population. Acromegaly is seen in 9 % of cases and has a greater female preponderance in MEN1 than in sporadic disease [105]. Nonsecreting adenomas, thyroid-stimulating hormone (TSH)-secreting adenomas, and adrenocorticotropic hormone (ACTH)-secreting adenomas do occur in MEN1 patients, in decreasing frequency [105]. MEN1-related pituitary tumors may exhibit immunoreactivity to several hormones, with a higher occurrence of somatolactotrophinomas [103]. Interestingly, plurihormonal expression is frequently observed in MEN1-associated pituitary tumors compared with sporadic pituitary tumors [105]. The MEN1 tumors seem to have a more aggressive behavior, to be more invasive (with infiltration of tumor cells into surrounding normal pituitary tissue) and more resistant to treatment, therefore requiring a long and careful life follow-up [106, 110]. However, no increased prevalence of pituitary carcinoma is observed in MEN1 (63). These tumors may occur in the pediatric population and may be the first and only manifestation of MEN1 for some years [106, 111].
20.2.3.1 Clinical Manifestations and Diagnosis
Clinical manifestations of MEN1-associated pituitary tumors are similar to those in sporadic pituitary tumors [103]. They depend on the hormone secreted (e.g., symptoms of increased prolactin, such as oligomenorrhea, galactorrhea, infertility in woman, impotence, and infertility in men; symptoms of acromegaly due to increased GH/IGF-I secretion; symptoms of Cushing’s disease due to increased cortisol secretion, etc.) and on the size of the tumor. Enlarging pituitary tumors may compress adjacent structures such as the optic chiasm or normal pituitary tissue and may cause visual disturbances and/or hypopituitarism [106].
20.2.3.2 Treatment of MEN1-Related Pituitary Tumors
Treatment of these tumors is similar to the one in patients with sporadic tumors and includes medical therapy (specifically dopamine agonists for prolactinoma or somatostatin analogues for GH-secreting adenoma together with surgical excision) and, if feasible, surgical excision, e.g., selective transsphenoidal tumor resection, with adjuvant radiotherapy in case of residual unresectable tumor tissue [105]. Noteworthy, as a result of the aggressive tumor behavior, MEN1 patients with pituitary tumors are less responsive to medical or surgical therapy, with a hormone normalization rate of 42 % compared with 90 % in patients with sporadic tumors, after therapy [103, 110, 112].
20.2.4 Other MEN1-Associated Tumors
MEN1 patients frequently develop tumors involving tissues other than the parathyroid glands, pancreas, and pituitary gland, such as neuroendocrine tumors (carcinoids), adrenal cortical tumors, thyroid tumors, facial angiofibromas, collagenomas, lipomatous tumors, and meningiomas [113, 114] (Table 20.1).
20.2.4.1 MEN1-Related Neuroendocrine Tumors (NETs, Carcinoids)
Clinical Manifestations and Diagnosis
NETs occur in ~ 3 % of MEN1 patients (Table 20.1), with the lungs, intestinal tract, or thymus being frequent sites of origin. In MEN1 patients, these tumors may be gender related, with a woman predominance for lung NETs and a male predominance for thymic NETs, cigarette smokers having an increased risk for developing these tumors [66, 115, 116]; moreover, there are no hormonal or biochemical abnormalities consistently reported in MEN1 patients with thymic or lung NETs. Importantly, MEN1-associated thymic NETs seem to be particularly aggressive, with a median survival time of approximately 9.5 years and with 70 % of patients dying as a direct result of the tumor [116].
Screening for these NETs is of major importance and their diagnosis depends on the imaging method used and the skills of the radiologist; CT and MRI are sensitive methods, whereas somatostatin-receptor scintigraphy may be useful, although there is still insufficient evidence to recommend its routine use [116, 117]. The current guidelines for the early detection of MEN1-related thymic and lung NETs recommend the use of CT or MRI imaging every 1–2 years (in favor of MRI, as repeated CT exposure to repeated doses of ionizing radiation may be harmful) [117, 118]. Noteworthy, thymic NETs have occurred also after prophylactic thymectomy, suggesting that surveillance imaging is still required [119] even after tumor excision.
Type II gastric NETs are associated with MEN1 and Zollinger-Ellison syndrome, are usually multiple and smaller than 1.5 cm, and may be detected incidentally during endoscopy in these patients [119].
Treatment of MEN1-Associated NETs
Surgical excision is the treatment of choice [119]. For unresectable or metastatic disease, treatment with somatostatin analogues, such as octreotide or lanreotide, has resulted in improvement in symptoms and tumor regression [119, 120]. In addition, chemotherapeutic agents (e.g., cisplatin, etoposide) or radiotherapy may be used in patients with aggressive tumors [121].
20.2.4.2 MEN1-Related Adrenal Tumors
Clinical Manifestations and Diagnosis
Adrenocortical tumors occur in up to 73 % of the patients with MEN1 (Table 20.1) [83, 122–124] and include adenomas, hyperplasia, cysts, or carcinomas. Most are nonfunctioning [122], whereas functional tumors are reported to occur in less than 10 % of patients, with primary hyperaldosteronism and ACTH-independent Cushing’s syndrome most commonly reported [122]. Pheochromocytomas are rarely described in MEN1 patients, whereas adrenocortical carcinoma is reported to occur in up to 13 % of MEN1 patients, in parallel with a tumor size of more than 1 cm [122].
Biochemical investigation (e.g., plasma renin and aldosterone concentrations, low-dose dexamethasone suppression test or 24-hour urine collection for free cortisol, urinary catecholamines and metanephrines, or plasma metanephrines) should be performed as routine for adrenal tumors. It is recommended that MEN1 patients with adrenal tumors should undergo an annual imaging screening (CT or MRI) of the adrenal glands (Table 20.2) [122, 124].
Table 20.2
Clinical manifestations in MEN2
Organ involved | Specific tumor/manifestation (prevalence by 40 years) | Clinical presentation | ||
|---|---|---|---|---|
MEN2 | MEN2A (>75 %) | Thyroid | MTC (90 %) | Neck mass or neck pain, diarrhea, flushing |
Adrenal | Pheochromocytomas (50 %) | Subtle symptoms, usually bilateral | ||
Parathyroid | Multigland adenoma/hyperplasia (20–30 %) | Usually asymptomatic | ||
Skin | CLA | Pruritic, lichenoid skin lesion in the upper portion of the back | ||
MEN2A genetically related | HSCR | Bowel enlargement and constipation/obstipation | ||
PTC | Concomitant PTC and MTC | |||
FMTC (10–20 %) | Thyroid | MTC (100 %) | ≥4 individuals with MTC in the same family in the absence of pheochromocytoma or parathyroid adenoma/hyperplasia | |
MEN2B (5 %) | Thyroid | MTC (90 %) | Very aggressive, usually metastatic | |
Adrenal | Pheochromocytomas (50 %) | Usually multiple/bilateral (50 %) | ||
Other | Mucosal neuromas, frequent | Tongue, palate, or pharynx; eyelid | ||
Ocular signs, frequent | Inability to cry tears, thickened corneal nerves, ptosis, eyelid eversion | |||
Diffuse ganglioneuromatosis of the GIT (40 %) | Abdominal distension, megacolon, constipation, or diarrhea | |||
Marfanoid habitus (75 %) | Associated with kyphoscoliosis or lordosis, joint laxity, decreased subcutaneous fat, proximal muscle wasting, weakness |
Treatment of MEN1-Related Adrenal Tumors
The treatment of MEN1-related adrenal tumors is similar to that for tumors occurring in the sporadic context. Surgical excision is recommended for adrenal tumors that are more than 4 cm in diameter, or less than 4 cm but with suspicious radiological features, or for tumors with significant growth over a 6-month interval [124].
20.2.4.3 MEN1-Related Central Nervous System (CNS) Tumors
20.2.4.4 MEN1-Related Thyroid Tumors
Thyroid adenomas, nodular goiter, and thyroid carcinoma have all been reported in more than 25 % of MEN1 patients. However, the occurrence of thyroid abnormalities in these patients may be incidental, as these disorders are also frequent in the general population. The treatment of MEN1-related thyroid tumors is similar to that for sporadic cases [50].
20.2.4.5 MEN1-Related Cutaneous Manifestations
Multiple subcutaneous lipomas are reported in more than 33 % of MEN1 patients (Table 20.1). Rarely, visceral, pleural, or retroperitoneal lipomas may also occur [50]. They are usually treated conservatively but may be excised for cosmetic reasons. Facial angiofibromas and collagenomas may occur in up to 88 % of MEN1 patients (Table 20.1), and usually no treatment is required [126].
20.2.5 Genetic Testing and Screening in MEN1
20.2.5.1 MEN1 Gene
The MEN1 gene is located on chromosome 11q13 and consists of 10 exons, which encode menin, a 610-amino-acid protein [127, 128], and a tumor suppressor that regulates transcription, genome stability, cell division, and proliferation [66, 129]. Inheritance of a germline MEN1 mutation predisposes an individual to develop a tumor that arises after an additional somatic mutation of the normal allele, which may be a point mutation or a deletion, leading to loss of heterozygosity (LOH) in the tumor DNA (the Knudson two-hit hypothesis) [66].
20.2.5.2 MEN1 Germline Mutations
Over 1,336 mutations of the MEN1gene have been characterized [66]. About 75 % of the MEN1 germline mutations are inactivating, consistent with those expected in a tumor suppressor gene. There are nine sites in the MEN1 gene accounting for over 20 % of all the germline mutations [66]. Noteworthy, it appears that there is no clear-cut correlation between MEN1 mutations and MEN1 clinical manifestations. The apparent lack of genotype-phenotype correlation, together with the wide diversity of mutations in the 1830-bp coding region of the MEN1 gene, renders mutational analysis more difficult [76, 129]. Importantly, more than 10 % of MEN1 germline mutations arise de novo [129]. Up to 25 % of MEN1 patients may not harbor germline mutations in the MEN1 gene coding region, probably related to whole or partial gene deletions, or mutations in the promoter or untranslated regions [129, 130]. Phenocopies may mimic MEN1 either by occurrence of a single sporadic endocrine tumor in a patient with familial MEN1 or occurrence of two endocrine abnormalities associated with different etiologies. Phenocopies arose in >5 % of MEN1 families and awareness of them is important in the clinical management of MEN1 and other hereditary disorders [131].
20.2.5.3 MEN1 Gene Polymorphisms
A gene is said to be polymorphic if more than one allele occupies that gene’s locus within a population [132]. Twenty-four different polymorphisms of the MEN1 gene have been reported [129]. The recognition of these polymorphisms is important as they may occasionally help in families without any MEN1 mutation identified [37].
20.2.5.4 MEN1 Tumor Somatic Mutations
More than 90 % of the tumors originating in MEN1 patients exhibit LOH on 11q13, consistent with the Knudson’s two-hit (second-hit) hypothesis [129]. Besides the LOH mechanism, the second-hit hypothesis may result also from intragenic deletions and point mutations.
20.2.5.5 MEN1 Variants
In rare families with MEN1 variants, only some of MEN1 characteristic clinical manifestations may be observed. Familial isolated hyperparathyroidism (FIHP) is a condition characterized by the development of parathyroid tumors as the sole endocrinopathy, being reported to date in 42 FIHP kindreds: 38 % developed missense mutations, and fewer than 31 % presented with nonsense or frameshift mutations [129, 134]. Another MEN1 variant is the Burin or prolactinomas variant, characterized by a high occurrence of prolactinomas and a low occurrence of gastrinomas [135–137] as result of nonsense mutations (Tyr312Stop and Arg460Stop). Another example is Tasmanian MEN1 kindred in whom the absence of somatotropinomas was observed as a result of a splice site mutation [138].
20.2.5.6 MEN1 Mutational Analysis in Clinical Practice
MEN1 mutational analysis is important for confirmation of the clinical diagnosis, for the identification of family members who require screening for tumor detection and appropriate treatment, as well as for the identification of the 50 % of family members who do not harbor the familial germline MEN1 mutation and can therefore be reassured [50].
MEN1 mutational analysis should be performed in the following circumstances [27, 139, 140]:
In an index case with ≥2 classic MEN1-associated endocrine tumors (i.e., parathyroid, pancreatic, or pituitary tumors)
In asymptomatic first-degree relative of a known MEN1 mutation carrier
In a first-degree relative of an MEN1 mutation carrier having symptoms, signs, biochemical, or radiological evidence for one or more MEN1-associated tumors
In patients with suspicious or atypical MEN1, including individuals with parathyroid adenomas diagnosed before the age of 30 years
In patients with multigland parathyroid disease, gastrinoma, or multiple pancreatic NETs at any age
In individuals who have two or more MEN1-associated tumors not part of the classical triad of parathyroid, pancreatic islet, and anterior pituitary tumors
In those individuals presenting at an early age with a single, apparently sporadic MEN1-associated tumor
20.2.5.7 MEN1 Tumors Identification
As MEN1 syndrome is associated with a complex spectrum of clinical, diagnostic, and therapeutic aspects, it is recommended for these patients and their families to be followed by teams with expertise in the management of MEN1, including an endocrinologist experienced in MEN syndromes [50]. As the MEN1 gene mutation appears to be nonpenetrant in individuals younger than 5 years, but with a >50 % penetrance by 20 years of age and >95 % by 40 years, a timely biochemical and imaging screening in asymptomatic members of MEN1 families is imperative, as earlier diagnosis and treatment may reduce the morbidity and mortality related to these tumors [5, 39, 66, 141].
PHPT-induced hypercalcemia is usually the first manifestation of MEN1 and therefore has become an easy biochemical screening investigation. In addition, hyperprolactinemia, usually asymptomatic, may represent the first manifestation in approximately 15 % of patients and may be easily detected [66]. PNETs may be detected in asymptomatic individuals by measuring fasting plasma concentrations of gastrin, pancreatic polypeptide, glucagon, and chromogranin A and by abdominal imaging [142, 143].
The latest guidelines [50] suggest that mutant MEN1 gene carriers should undergo biochemical screening (including evaluation of serum calcium, PTH, gastrin, insulin with a fasting glucose, glucagon, VIP, pancreatic polypeptide, chromogranin A, prolactin, and IGF-I) at least once a year. Baseline pituitary and abdominal imaging (e.g., MRI or CT) should be performed and then repeated at 1–3 years’ interval, as well as imaging for thymic and bronchial carcinoids using CT or MRI every 1–2 years. Screening should possibly commence in early childhood (e.g., by the age of 5 years) and should be repeated throughout life [50].
20.3 Multiple Endocrine Neoplasia Type 2 (MEN2, Sipple Syndrome)
Type 2 multiple endocrine neoplasia (MEN2) is a rare familial cancer syndrome caused by mutations in the RET proto-oncogene. The association between thyroid cancer and pheochromocytoma was first described by Sipple in 1961 [144]. In 1965, the thyroid cancer occurring in association with pheochromocytoma was defined as medullary thyroid carcinoma (MTC), characterized by stromal amyloid. Since 1968, the familial constellation of MTC in conjunction with pheochromocytoma and with parathyroid hyperplasia was recognized as MEN2. Although patients with mucosal neuromas were already identified, the distinction between MEN2A and MEN2B subtypes was only in 1975 [145].
MEN2 is an autosomal dominant syndrome classified into main two variants: MEN2A and MEN2B [146]. All variants of MEN2 show a high penetrance for MTC, which is diagnosed in as much as 90 % of MEN2 carriers (as a palpable thyroid nodule or an increase in blood calcitonin levels, CLT) [146, 147].
All MEN2 subtypes are caused by a germline mutation in the RET proto-oncogene and specifically with selected RET codon mutations [148, 149].
20.3.1 The Clinical Presentation of MEN2 Syndromes (Table 20.2)
20.3.1.1 Clinical Presentation of MEN2A
MEN2A is characterized by the development of MTC in 90 % of adult gene carriers, unilateral or bilateral pheochromocytoma in 50 %, and multigland parathyroid adenoma/hyperplasia with primary hyperparathyroidism (PHPT) in 20–30 % [150–152]. MEN2A accounts for over 75 % of MEN2 [153] and may present as rare variants including familial MTC (FMTC) [151, 154], MEN2A with cutaneous lichen amyloidosis [155, 156], and MEN2A or FMTC with Hirschsprung’s disease [157].
MEN2A-Related MTC
Hereditary MTC is a rare calcitonin (CLT)-producing tumor developing from the parafollicular or C cells of the thyroid gland [158, 159]. Multifocal C-cell hyperplasia (CCH) is the precursor lesion to hereditary MTC, and the age of transformation from CCH to microscopic MTC varies with different germline RET mutations and may take years [160, 161]. MTC is generally the first manifestation of MEN2A, and the patients typically present with a neck mass or neck pain, usually before age 35 years, with cervical lymph node metastases in up to 70 % [162]. Diarrhea, the most frequent systemic manifestation (~30 % of patients with advanced MTC), occurs in affected individuals with a plasma calcitonin concentration of >10 ng/mL and implies a poor prognosis [163]. Approximately 10 % of MTC patients have flushing often induced by alcohol ingestion, calcium infusion, or pentagastrin injection [164]. All individuals with an MTC-predisposing mutation who have not undergone prophylactic thyroidectomy demonstrate biochemical evidence of MTC by age 35 years [147]. Metastatic spread may occur locally (to the central, lateral, cervical, and mediastinal lymph nodes) or distantly (to the lungs, liver, or bones) [165].
MEN2A-Related Pheochromocytomas
Pheochromocytomas usually present concomitantly or after MTC; however, they may be the presenting symptom in up to 27 % of individuals with MEN2A [166–168]. MEN2-related pheochromocytomas are usually diagnosed earlier, have subtler symptoms, and are more likely to be bilateral than sporadic tumors [169, 170]. Pheochromocytomas in MEN2 patients entail a distinct biochemical phenotype in that they consistently produce epinephrine or epinephrine and norepinephrine [171]. Malignant transformation was reported in ~ 4 % of cases [172].
MEN2A-Related Primary Hyperparathyroidism (PHPT)
MEN2A-related PHPT is usually mild, being diagnosed many years after the diagnosis of MTC, with an average age of onset ~ 38 years [173]. Most individuals are asymptomatic; however, hypercalciuria with renal calculi may occur. In case of long-standing or underdiagnosed disease, hypercalcemia-related symptoms may develop and become severe, in parallel with disease progression [174].
20.3.1.2 Clinical Presentation of FMTC
FMTC is historically diagnosed in families with four or more cases of MTC in the absence of pheochromocytoma or parathyroid adenoma/hyperplasia [153, 154] and comprises approximately 10–20 % of cases of MEN2. Because RET accounts for all clinical subtypes of MEN2, FMTC may be viewed as MEN2A with reduced organ-specific penetrance. The age of onset of FMTC is later, and the penetrance of MTC is lower than what is observed in MEN2A and MEN2B [177, 178].
20.3.1.3 Clinical Presentation of MEN2A Genetically Related (Allelic) Disorders
Hirschsprung’s Disease (HSCR)
HSCR is a genetic disorder characterized by aganglionosis of the gut, likely due to absent gut ganglia and typically resulting in bowel enlargement and constipation/obstipation in neonates [179]. Up to 50 % of familial cases and up to 35 % of simplex cases (i.e., a single occurrence in a family) of HSCR are caused by germline loss-of-function mutations in the RET proto-oncogene [180, 181].
Papillary Thyroid Carcinoma (PTC)
20.3.1.4 Clinical Presentation of MEN2B
MEN2B comprises approximately 5 % of cases of MEN2, and it is the most aggressive variant of MEN2 syndrome, characterized by MTC and pheochromocytoma, together with a marfanoid habitus and mucosal (lips and tongue) and intestinal ganglioneuromatosis, but not PHPT [186, 187].
MEN2B-Related MTC
MEN2B-related MTC is the most aggressive form of MTC in all affected individuals and metastasizes in all MEN2B carriers who do not undergo thyroidectomy at an early age (<1 year). Before intervention with early prophylactic thyroidectomy, the average age of death in individuals with MEN2B was 21 years [188].
MEN2B-Related Pheochromocytomas
Pheochromocytomas occur in 50 % of individuals with MEN2B; approximately half are multiple and often bilateral [189]. Most commonly, these patients complain of palpitations, anxiety spells, or headache early in their disease; however, in many patients whose tumors were found by family screening, there are no symptoms. Hypertension spells may not be noted until a crisis is induced by an operation or delivery [190].
MEN2B-Related Other Clinical Presentations
Mucosal neuromas (Figure 20.1) may be identified in early childhood in MEN2B carriers and develop on the anterior dorsal surface of the tongue, palate, or pharynx, together with a distinctive facial appearance. The lips become prominent (or “blubbery”) over time as a result of submucosal nodules present on the vermilion border of the lips. Eyelid neuromas may cause thickening and even eversion of the upper eyelid margins [191].
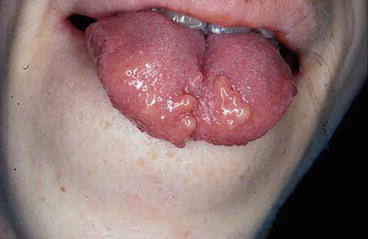
Fig. 20.1
MEN2B-associated oral mucosal neuromas
Ocular Signs of MEN2B: children with MEN2B have the inability to cry tears, and this is an important clinical symptom for early diagnosis preceding the development of metastatic MTC [192]. Other ocular signs develop during childhood and include prominent thickened corneal nerves (seen by slit lamp examination) [193, 194], eyelid neuromas, lid margin eversion or thickening, subconjunctival neuromas, or ptosis [195–197]. There are usually no fundal findings on examination, but in patients presenting already with MTC, choroidal metastases have been observed [198].
Diffuse ganglioneuromatosis of the gastrointestinal tract is reported in ~ 40 % of affected individuals, and it is associated with early childhood symptoms of aganglionic megacolon [199].
Marfanoid habitus, often with kyphoscoliosis or lordosis, joint laxity, decreased subcutaneous fat, proximal muscle wasting, and weakness, can also be seen in ~ 75 % of affected individuals [200].
20.3.2 The Diagnosis of MEN2 Syndrome Manifestations
20.3.2.1 MTC Diagnosis
CLT values (basal or stimulated by pentagastrin, calcium, or both) are nearly always elevated in MTC [201], being a specific and a sensitive marker, and elevation after surgery suggests persistent or recurrent disease [202]. In provocative testing, plasma CLT concentrations are measured before and 2 and 5 min after intravenous administration of the CLT secretagogue. Reference levels for basal calcitonin vary across laboratories, and usually levels of <10 pg/mL for adult males and <5 pg/mL for adult females are typically considered normal. Furthermore, a basal or stimulated calcitonin level of ≥100 pg/mL is considered an indication for surgery [203, 204]. Preoperative calcitonin testing may be useful for assessing CCH (C-cell hyperplasia) or tumor spread and should be considered when deciding the extent of surgery for MEN2A MTC [205].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


