less than 3 seconds can be spontaneous or induced by photic stimulation (2,5). Discharges featuring multiple generalized spikes and waves often increase in frequency during nonrapid eye movement (NREM) sleep (2,5).
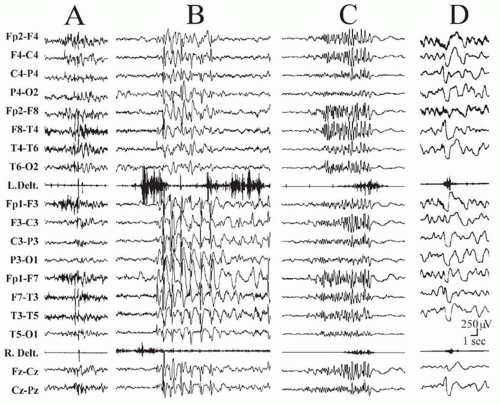 Figure 26.1 Types of brief seizures that clinically manifest as body jerks, shown by their neurophysiologic correlates. A: Myoclonic seizure; electromyographic (EMG) burst lasts less than 100 milliseconds and is time-locked to a polyspike-and-wave discharge. B: Atonic seizure; a spike-and-wave discharge is accompanied by an EMG silent period lasting as long as the discharge on scalp electroencephalography. Note the progressive onset of the silent period and the transient resumption of muscle tone, resembling a myoclonic jerk, interrupting the EMG silence. C: Brief tonic seizure; a polyspike discharge is associated with a bilateral tonic contraction of both deltoid muscles lasting about 5 seconds, that begins with a crescendo pattern. D: Epileptic spasm; a brief (approximately 500 milliseconds), diamondshaped EMG burst correlated with a high-amplitude, diffuse, polyphasic slow transient on EEG channels. |
channel may represent the pathophysiologic substrate for at least some of the nonprogressive myoclonic epilepsies. On the other hand, the progressive myoclonic epilepsies (PMEs), which are also genetically determined, follow an autosomal recessive inheritance and have been related to gene defects causing abnormal deposit material to accumulate in various organs including the brain.
TABLE 26.1 CLASSIFICATION OF MYOCLONIC EPILEPSIES BY AGE OF ONSET | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
muscle contraction and occur in clusters. Polygraphic EEG-EMG recordings easily clarify the clinical picture. Benign nonepileptic myoclonus of early infancy should be considered in the differential diagnosis of developmentally normal children who present with clusters of jerky movements of the limbs, nodding, or axial shudder but have normal interictal and ictal EEG findings (44,45). In such cases, polygraphic recordings show a tonic, rather than a myoclonic, contraction lasting 0.5 to 3 seconds (44).
with parietal accentuation and occipital 4-Hz rhythms, constantly blocked by eye opening (6). Variable lateralization of paroxysmal bursts is possible, although a consistently localized focus is unusual (50). A subset of patients are photosensitive.
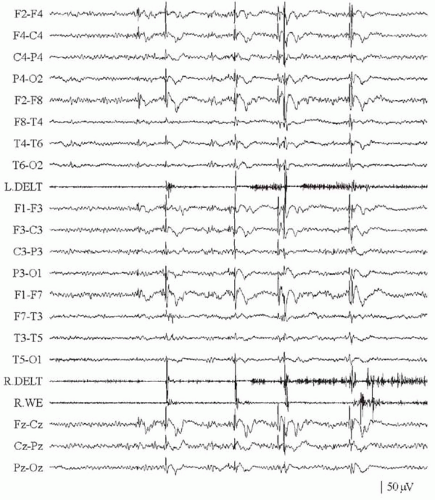 Figure 26.2 Surface electroencephalographic (EEG)-electromyographic (EMG) polygraphic recording of a 13-year-old boy with myoclonicastatic epilepsy. Generalized myoclonic jerks are visible as high-amplitude myoclonic potentials on the EMG channels accompanied by a diffuse discharge of spike-wave complexes. A clear time-locked correlation is observed between the EEG spikes and the myoclonic EMG potentials. L.DELT = left deltoid muscle; R.DELT = right deltoid muscle; R.WE = right wrist extensor. |
but not incompatible with MAE. Other childhood epilepsies with predominant myoclonic attacks remain difficult to classify and in some cases may belong to the spectrum of MAE. It is difficult to subsume all myoclonic patients into single, well-defined syndromic entities (51). Of more use is to consider idiopathic epilepsies with prominent myoclonic seizures, such as BMEI, MAE, and other unclassifiable cases, as part of a continuum with different degrees of severity.
motor cortex of one hemisphere and either hemisphere can be activated in subsequent discharges (1). Multifocal erratic myoclonic jerks can involve the hands and face (5). Their origin remains speculative, as back-averaging studies did not produce any cortical transient time-locked to the jerks (1).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


