Toxic and Nutritional Disorders
John H. Menkes
TOXIC DISORDERS
Neurotoxins are endogenous or environmental. Various sections of this text consider the endogenous neurotoxins, notably the excitatory neurotransmitters. These substances are implicated in cell death from asphyxia and hypoglycemia, following uncontrolled seizures, and possibly in some of the heredodegenerative disorders. This chapter discusses environmental toxins. Almost all environmental toxins induce neurologic symptoms if ingested in sufficient quantity. This chapter considers only those that are encountered in clinical practice commonly and for which the nervous system is a primary site of action. They are discussed in ascending order of their chemical complexity.
Drug intoxications that are encountered less commonly by the practitioner are not considered in detail. The reader is referred to Table 10.1 (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24) for a summary of their neurologic complications and treatment.
In addition, the reader is directed to the comprehensive text on medical toxicology edited by Ellenhorn and colleagues (25), and a book on neurotoxicology edited by Spencer and coworkers (26).
The incidence of the various drug intoxications in an Australian pediatric population during the years 1977 through 1981 is depicted in Table 10.2 (27). A series compiled from the Children’s Hospital of the University of Antwerp for the years 1980 through 1988 shows a similar distribution (28).
Metallic Toxins
Lead
Although some of the toxic actions of lead have been known since antiquity, the understanding of the clinical picture of lead poisoning is based on the classic studies of Tanquerel des Planches (29), Blackfan (30), and Holt (31). As a result of the considerable advances in the prevention of lead exposure, cases of acute lead encephalopathy have all but disappeared from the United States. Nevertheless, children with increased blood lead levels are still being encountered with considerable frequency, and the effect of elevated blood lead levels on neurocognitive and neurobehavioral function has been the subject of considerable recent controversy.
Pharmacology
One of the major sources of lead continues to be exterior and, to a lesser extent, interior lead-based paint, which remains as an environmental hazard in some 3.8 million homes in the United States housing children 7 years of age or younger (32). Lead can be absorbed by water with a low mineral content as it passes through lead pipes or lead-soldered copper pipes. It also can be ingested when water is boiled in soldered electric kettles. Until the 1990s, organic lead also was ingested from roadside dust contaminated with automobile emissions, or by habitual sniffing of leaded gasoline. Less common sources of lead include lead arsenate insecticides; lead solder, which in the United States has been virtually eliminated from the seams of food and beverage cans; ancient pewter; imperfectly glazed ceramics; painted jewelry; and eye shadow, which in some developing countries is applied to infant boys and girls. One form of lead poisoning, once relatively common in nursing infants, was caused by prolonged use of lead nipple shields.
The gastrointestinal tract of infants and children absorbs approximately 40% of ingested lead, with the remainder being excreted. Approximately 90% of inhaled lead is absorbed. Iron deficiency and constipation encourage absorption; diarrhea has the reverse effect. Lead and calcium compete for a common transport mechanism; reduced calcium intake and low serum levels of 25-hydroxy vitamin D1 increase lead absorption. The vitamin D receptor gene is involved in calcium absorption and, if lead is present, will also absorb lead, and genotype variants of the vitamin D receptor gene may influence the degree of lead absorption (33). After absorption, lead is distributed into three compartments: Some enters a rapidly exchangeable pool in the liver and kidney and from there binds to a low-molecular-weight protein of erythrocytes (34); some enters a rapidly exchangeable pool in soft tissue and is loosely bound to bone; and the major portion (60% to 90%) is
tightly bound as the insoluble and nontoxic lead triphosphate in the skeleton, notably in the epiphyseal portion of growing bone. Lead also is deposited in hair and nails and crosses the placenta. Only a small amount of lead enters the brain; the major portion is found in gray matter and the basal ganglia (35,36). High phosphate and high vitamin D intakes favor skeletal storage of lead, whereas parathormone, low phosphate intake, and acidosis promote its release into the bloodstream. Almost all the absorbed lead is excreted in urine; fecal lead represents the unabsorbed fraction. The half-life of lead in blood is 1 to 2 months; it is 20 to 30 years in bone (36). Excretion is so slow that even a slight increase in the average daily intake above 0.3 mg/day, the maximum permissible level for adults, can ultimately induce poisoning.
tightly bound as the insoluble and nontoxic lead triphosphate in the skeleton, notably in the epiphyseal portion of growing bone. Lead also is deposited in hair and nails and crosses the placenta. Only a small amount of lead enters the brain; the major portion is found in gray matter and the basal ganglia (35,36). High phosphate and high vitamin D intakes favor skeletal storage of lead, whereas parathormone, low phosphate intake, and acidosis promote its release into the bloodstream. Almost all the absorbed lead is excreted in urine; fecal lead represents the unabsorbed fraction. The half-life of lead in blood is 1 to 2 months; it is 20 to 30 years in bone (36). Excretion is so slow that even a slight increase in the average daily intake above 0.3 mg/day, the maximum permissible level for adults, can ultimately induce poisoning.
TABLE 10.1 Neurotoxic Complications of Some Drugs | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 10.2 Agents Accidentally Ingested by Children (0 Through 12 Years) Resulting in Hospital Admission | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
The levels of lead in blood and urine are an index of the degree of exposure to the toxin. Although blood lead levels below 10 μg/dL are currently considered acceptable, δ-aminolevulinic acid dehydratase (ALAD) activity is significantly inhibited at blood lead levels of 5 to 10 μg/dL (37), and abnormalities in auditory and visual-evoked potentials have been demonstrated at levels below 10 μg/dl. (38). Measurement of hair lead is an unreliable indicator of the severity of lead exposure, whereas the reliability of tooth lead content is still under debate (39). As yet, the threshold level for neurotoxicity is unknown. The American Academy of Pediatrics has stated that concentrations of 25 μg/dL or higher are certain to be toxic, and the average blood lead level in American children is 2.6 μg/dL (40).
Pathology
Almost all organs are affected by lead poisoning, with damage caused by heavy metal inhibition of numerous sulfhydryl enzymes and its ability to substitute for calcium,
and perhaps zinc (38). One of the earliest manifestations of lead intoxication is anemia, induced by a disturbance in heme synthesis and by shortening of red cell life span. Lead impairs heme synthesis at several points, with globin synthesis also being disrupted at higher lead levels (36,41).
and perhaps zinc (38). One of the earliest manifestations of lead intoxication is anemia, induced by a disturbance in heme synthesis and by shortening of red cell life span. Lead impairs heme synthesis at several points, with globin synthesis also being disrupted at higher lead levels (36,41).
Within the central nervous system (CNS), lead acts primarily on capillary endothelium. It causes extravasation of plasma, followed by endothelial, microglial, and astrocytic proliferation and widespread interstitial edema (42). Neuronal degeneration is also widespread, with the neocortex and cerebellum being affected most prominently; neocortical degeneration also can reflect agonal anoxia, rather than a direct effect of lead. Experimental work suggests that the immature cerebral vasculature is particularly sensitive to lead, and that neurotoxicity is the result of multifactorial events. Lead interrupts the cellular calcium pump in mitochondria with a resultant calcium-mediated cell death in the cerebrovascular endothelium (43). Chronic exposure to lead interferes with calmodulin-dependent processes resulting in abnormal post-translational modifications of cytoskeletal proteins. In addition, lead inhibits acetylcholine release and enhances the release of dopamine and norepinephrine (44). These multiple neurotoxic effects of lead are reviewed by Lidsky and Schneider (38).
In peripheral nerves, axonal degeneration occurs with loss of large myelinated fibers, but with no demyelination (45). Occasionally, the anterior horn cells also are degenerated. Chronic, low-level exposure, as is seen in industrial workers, can produce paranodal demyelination and internodal remyelination (46). No explanation exists for the preferential involvement of nerves in the upper extremity.
Pathologic changes within other organs, notably the liver and kidney, have been described in a classic publication by Blackman (47).
TABLE 10.3 Signs and Symptoms of Overt Lead Poisoning in Children (Chicago, 1955–1957) | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||
Clinical Manifestations
Neurologic symptoms of lead poisoning form a continuum from the most minor to the most severe (48). In the classic form of severe lead encephalopathy, symptoms progress insidiously, first becoming apparent in late summer in some 80% of cases. Before then, there is usually a prodromal period of several weeks or months during which the child is pale, irritable, listless, and without appetite. Epigastric pain, vomiting, and constipation are common. These nonspecific symptoms are often interrupted by the sudden onset of a series of generalized convulsions or by depression of consciousness (Table 10.3) (49,50).
Seizures are common and resist anticonvulsant therapy. They can be followed by hemiplegia or other neurologic sequelae.
The child with lead encephalopathy demonstrates increased intracranial pressure. Less frequent is cerebellar ataxia and palsies of the sixth and seventh cranial nerves. Nuchal rigidity is fairly common and can be related to tonsillar herniation. Patients in some parts of the world have a high incidence of optic neuritis with profound vision loss, acute communicating hydrocephalus, and tremors (51).
In children, peripheral neuritis caused by lead poisoning is rare. When seen, it almost invariably occurs in subjects with sickle cell anemia (52).
Anemia and deranged renal tubular function usually accompany the neurologic symptoms. Red cells in peripheral blood and bone marrow can show basophilic stippling, and the presence of a large number of such cells is almost invariable in chronic poisoning caused by inorganic lead, although not in poisoning with organic (tetraethyl) lead. Basophilic stippling, however, is not specific for lead intoxication.
Radiologic findings in acute lead encephalopathy are characteristic (53). Most prominent is the presence of a dense, radiopaque band at the metaphyses of numerous long bones. Less commonly, radiopaque particles are
found within the gastrointestinal tract as evidence of recent plaster ingestion.
found within the gastrointestinal tract as evidence of recent plaster ingestion.
Although lead encephalopathy and peripheral neuritis represent the classic and severe form of lead intoxication, neurobehavioral symptoms as a consequence of lesser degrees of exposure are now far more common, and have, in the last few years, become a contentious issue for health care workers.
A large number of epidemiologic studies designed to examine the effects of low-level lead exposure on the nervous system differ with respect to the measures selected to reflect the amount of lead to which the developing brain has been exposed. They also differ in the areas of cognitive and neurologic performance chosen to measure the effects of lead intoxication. In almost every study, the results have been confounded by the association between increased lead intake and disadvantaged family and home environment. Smith (54) and Pocock and colleagues (39) have extensively and dispassionately reviewed these issues.
I concur with their conclusions that from the current state of knowledge one is unable to say whether low-level lead exposure affects performance or behavior of children, or whether a reverse causality exists (i.e., children with lower IQs or aberrant behavior patterns have increased lead uptake). In either case, in cases in which effects of environmental lead are found, they are likely to be small and are overshadowed by the effects that result from the child’s socioeconomic status, quality of home environment, and parental intelligence (39,54,55). In addition, the effects of genetic and dietary factors on lead absorption and neurocognition have not been clarified. Most significantly, a blood lead level below which lead exposure has absolutely no effect on neurocognitive function has not yet been established (56). Finally, we also do not know what role, if any, intrauterine exposure to lead plays in inducing cognitive deficits (39,55). Thus, in the Australian study of Baghurst and coworkers, no significant relation existed between IQ at 57 months of age and antenatal or perinatal blood lead concentrations (55).
A further discussion on the interrelation between lead exposure and cognitive function can be found in Chapter 18.
Diagnosis
Even though lead encephalopathy has become extremely rare, the appearance of seizures and increased intracranial pressure in a young child should always suggest this condition. Posterior fossa tumors rarely produce seizures, and tumors of the cerebral hemispheres are rare in toddlers. Lead lines on radiographic examination and basophilic stippling in a blood smear offer a presumptive diagnosis even in the absence of a history of pica.
The differential diagnosis between a sickle cell crisis that produces neurologic symptoms and lead poisoning can be difficult because both can produce cramping abdominal pains and seizures. Generally, neurologic signs in sickle cell crisis are caused by cerebrovascular occlusion and thus result in lateralizing findings, which are generally absent in lead encephalopathy.
Currently, the most widely used test to detect chronic low-grade exposure to lead is the whole blood lead level, with the analysis being performed by atomic absorption spectrophotometry. Capillary blood can be used for diagnosis and surveillance (57), and for screening purposes, a filter paper blood test has been found to be reliable (58). An increased hair lead level has been suggested as an indicator for the timing of increased exposure to lead. This measurement, however, is unreliable in that hair lead concentrations can vary in different hairs from the same individual. It also is difficult to remove surface contaminants without removing lead from within the hair. The lead contact of teeth has been used as an indicator of cumulative lead exposure, but this test is obviously impractical for the routine diagnosis of lead poisoning (59).
Because peripheral nerve conduction times are slowed in many youngsters with even inapparent neuropathy, peripheral nerve conduction studies can provide an ancillary indication of subclinical lead intoxication (52).
Treatment and Prognosis
Because recurrent exposure is common, and because neurologic damage caused by lead poisoning is probably progressive rather than all or none, the source of the ingested lead must be ascertained and eliminated before the child returns home from the hospital. The recommended practice is to forego treatment for children whose blood lead level is less than 25 μg/dL. Children with blood lead levels between 25 and 45 μg/dL require their immediate removal from lead exposure, but neither chelation therapy nor dietary calcium supplementation is indicated (60,61). Chelation therapy is indicated for patients with blood lead levels over 45 μg/dL. Two parenteral and two oral chelation agents are currently available. The former are dimercaprol (BAL) and calcium disodium ethylenediaminetetraacetic acid (EDTA). Oral chelation agents are succimer, a water-soluble analogue of BAL, and D-penicillamine. A full discussion of dosages, duration of therapy, and adverse reactions is beyond the scope of this text. They are detailed in the treatment guidelines prepared by the American Academy of Pediatrics, Committee on Drugs (62), in a publication by Piomelli (63), and in a review by Henretig (64). Case management and pitfalls in the treatment of children with severe lead poisoning are discussed by Friedman and Weinberger (65).
Even with the best therapy, the prognosis for complete recovery in children with lead encephalopathy is not good, and by the time symptoms become overt, lasting CNS damage has already been incurred.
Thallium
Children can accidentally ingest thallium-containing pesticides. How the metal damages the nervous system is still unknown (66). One likely possibility is that thallium replaces potassium in the activation of potassium-dependent enzymes, notably pyruvate kinase and succinate dehydrogenase. In fatal cases, one sees white matter edema and degenerative changes in the nerve cells of the cerebral and cerebellar cortex, hypothalamus, olivary nuclei, and the corpus striatum. In peripheral nerves, there is a loss of myelinated fibers and degeneration of the central axons of sensory ganglion cells (67).
Depending on the dose ingested, intoxication can be fatal during the acute phase by causing renal and cardiac failure. In less severe intoxications, neurologic symptoms appear 2 to 6 days after ingestion, including a motor neuropathy that principally affects the lower extremities. Hair fibers in the growing stage, which constitute between 80% and 90% of scalp hair, and large-diameter fibers are most vulnerable to thallium intoxication (68). Subsequently, cerebellar ataxia can develop, accompanied by somnolence or seizures. A characteristic depilation resembling that seen after vincristine therapy begins within 2 weeks. Less commonly, there are retrobulbar neuritis and cranial nerve palsies (69).
Approximately one-half of the survivors have persistent neurologic deficits, most commonly mental retardation and ataxia. The diagnosis suggested by neurologic symptoms in the presence of depilation can be confirmed by the detection of thallium in urine using atomic absorption spectroscopy and emission spectrography (70).
Chelating agents such as diethyl dithiocarbamate or dimercaptosuccinic acid are ineffective in the treatment of thallium poisoning. Although they increase thallium excretion, the chelate formed is lipophilic and crosses the blood–brain barrier. As a consequence, neurologic symptoms worsen (71). Prussian blue, however, does have some value, having induced clinical improvement in a number of patients (72).
Arsenic
In the Western world, arsenic poisoning in the pediatric population most commonly results from the accidental ingestion of pesticides, notably ant poison (73). Since 1980, arsenic toxicity has become a major health problem in some developing countries with arsenic contaminating drinking water obtained from wells. The metal can arise from natural geological sources such as iron-pyrite as a result of overexploitation of ground water as well as from contamination induced by mining and other industrial processes. Arsenic poisoning is particularly common in Bangladesh and West Bengal, where up to 6% of children demonstrate symptoms of arsenic poisoning (74,75). The pathologic changes in the brain have been reviewed in a classic paper by Russell (76). They include generalized edema and pericapillary hemorrhages within white matter, with foci of demyelination. Nerve biopsies show axonal degeneration (77). The toxicity of arsenic probably lies in its ability to inactivate up to 200 sulfhydryl enzymes, notably those involved in the cellular energy pathways and DNA synthesis and repair (74,78).
Symptoms of arsenic poisoning in the pediatric population are more likely to be gastrointestinal than dermatological (pigmentation and keratosis) or neurologic (75,79). After acute arsenic ingestion, they can take the form of a violent attack of vomiting and bloody diarrhea. Chronic arsenic intake induces intermittent gastrointestinal upsets. In children, anemia is not unusual. Neurologic symptoms are mainly those of a peripheral sensory neuritis; numbness and severe paresthesias of the distal portion of the extremities predominate. Distal weakness of the lower extremities follows and can extend to involve the upper extremities.
Mercury
Mercury poisoning can result from exposure to elemental mercury in the form of its vapor, inorganic mercury salts, and organic mercury, most commonly as methylmercury. Because of their ability to affect neuronal cell division and migration, organic mercury compounds are highly toxic to the fetus and the early developing postnatal brain.
Mercury vapor poisoning has been reported to induce seizures. Pink disease (acrodynia), once relatively common owing to ingestion of calomel (mercuric chloride) teething powders (82), is now much rarer but has been observed after exposure to latex paint, which up to 1990 contained phenylmercuric acetate. Mercury is released from surfaces coated with this paint after it has dried (83). Only a small proportion of exposed infants and young children become symptomatic. Clinical manifestations are marked by erythema, most intense over the fingertips and toes; profound weakness of all extremities; hyperesthesia; extreme irritability; and changes in consciousness.
Intoxication with organic mercury compounds derived from the fungicides or methylmercury was responsible for outbreaks of poisoning in Iraq (84), whereas ingestion of methylmercury-contaminated fish resulted in Minamata disease, an encephalopathy encountered along Minamata Bay in Japan. The neurologic picture of organic mercury poisoning in children is complex. Tingling and other sensory disturbances and progressive visual impairment, taking the form of constriction of visual fields,
tunnel vision, or complete blindness, are almost invariable. Ataxia, tremor, impaired hearing, and mental deterioration also are encountered. A variety of involuntary movements are less common (85). Magnetic resonance imaging (MRI) shows atrophy of the ventral portion of the calcarine sulcus (86). Although neurological degeneration was progressive in the Japanese epidemic, the more recently affected Iraqi children have had considerable functional recovery (84,87).
tunnel vision, or complete blindness, are almost invariable. Ataxia, tremor, impaired hearing, and mental deterioration also are encountered. A variety of involuntary movements are less common (85). Magnetic resonance imaging (MRI) shows atrophy of the ventral portion of the calcarine sulcus (86). Although neurological degeneration was progressive in the Japanese epidemic, the more recently affected Iraqi children have had considerable functional recovery (84,87).
Methylmercury compounds also have been held responsible for congenital mercury poisoning. Affected infants show tremors, hypotonia, myoclonic jerks, and microcephaly (88,89). Researchers have studied the role of prenatal and early postnatal exposure to methylmercury derived from fish and other seafood—where it is found coordinated with an aliphatic thiol, most likely cysteine—on early development and neurocognitive functions.Whereas studies from the Faeroe Islands and New Zealand (90) have shown subtle but significant adverse effects in school-age children accompanied by abnormalities of the brainstem auditory-evoked potentials (91), the more recently conducted studies from the Seychelles Islands found no developmental effect of prenatal mercury exposure (92). The different outcomes could in part reflect the different concentrations of methylmercury consumed in these areas, since in the Seychelles, seafood has lower concentrations of methylmercury than in the other two regions (92). In addition, the Faroese population was exposed to polychlorinated biphenyls (PCP) and other organic pollutants that can act synergistically with mercury (92a).
Thimerosal, a mercury-containing compound that is degraded to ethylmercury and until the early 1990s was used as a preservative in various vaccines, has been implicated in neurodevelopmental disorders, particularly regressive autism (see Chapter 18). At present, the body of existing data is not consistent with an association between thimerosal-containing vaccines and autism or neurodevelopmental disorders (93,93a,94,94a).
The neuropathology of organic mercury poisoning in children has been described by Takeuchi and coworkers (95). It consists of focal cerebral and cerebellar atrophy with the axonal retraction bulbs (torpedoes) on Purkinje cells resembling those seen in Menkes disease (see Chapter 1).
Manganese
Whereas in the adult population manganese intoxication is mainly incurred as a consequence of chronic inhalation of the metal dust by welders or miners, in the pediatric age group, this condition is seen in patients who have been receiving long-term parenteral nutrition, particularly in the presence of chronic hepatic disease. Manganese exposure leads to a selective dopaminergic dysfunction, neuronal loss, and gliosis in the basal ganglia. Astrocytes appear to be particularly vulnerable in that they have a specific transport system for manganese facilitating its uptake and sequestration in mitochondria (98). The condition results in a variety of movement disorders, notably a parkinsonian picture, seizures, or myoclonus (99). The MRI studies are characteristic. They demonstrate hyperintensities in the globus pallidus on T1-weighted images (100). Chelation treatment is said to be beneficial and results in improvement of symptoms and the neuroradiologic abnormalities.
Aluminum
Aluminum intoxication, usually caused by the ingestion of aluminum hydroxide, has been implicated in the pathogenesis of progressive dementia and seizures in children with chronic renal failure, whether or not they are undergoing maintenance dialysis. Aluminum intoxication is discussed more fully in Chapter 17.
Organic Toxins
Carbon Monoxide
Carbon monoxide poisoning is one of the major causes of death caused by toxins. In infants and small children, the majority of cases results from faulty home heaters or, as in the Orient, the use of anthracite coal briquettes. The brain and the myocardium are most vulnerable to carbon monoxide. The principal effect of carbon monoxide is to interfere with tissue oxygenation by reducing the capacity of blood to transport oxygen and by shifting the blood oxyhemoglobin saturation curve, thus requiring blood oxygen tension to decrease to lower than normal values before a given amount of oxygen is released from hemoglobin. Carbon monoxide also reduces oxygen delivery to tissue by reducing cardiac output. In addition, carbon monoxide binds directly to iron-rich brain regions, notably the globus pallidus and the pars reticulata of the substantia nigra (101).
Neurological symptoms of carbon monoxide toxicity include irritability, lethargy, and coma. In the Korean series of Kim and Coe, 14% of children presented with convulsions, in 13% there was neck stiffness, and 3% had blindness on admission. Computed tomographic scans showed diffuse cerebral edema and low-density areas in the basal ganglia, notably, the globus pallidus (102). On T2-weighted MRI, increased signal is seen in the basal ganglia and white matter (102a). Chronic carbon monoxide poisoning can result from poorly ventilated heating systems and
inadequately ventilated housing. Symptoms include hyperactivity, sleepiness, and headaches (103).
inadequately ventilated housing. Symptoms include hyperactivity, sleepiness, and headaches (103).
After apparent recovery, some 10% to 30% of patients, most of them of advanced age, who have had moderate to severe carbon monoxide poisoning develop delayed neurologic sequelae some 3 to 240 days after intoxication (104,105,106). The sequelae include a variety of extrapyramidal symptoms, notably tremor, rigidity, and choreoathetosis. Additionally, Kim and Coe encountered mental retardation, seizures, mutism, hemiparesis, and paraplegia (102). MRI in such patients shows a progressive demyelinating process (107). The underlying mechanism for the delayed neurologic symptoms is currently believed to involve carbon monoxide–mediated brain lipid peroxidation (106,108).
The pros and cons for the treatment of children suffering from carbon monoxide poisoning with hyperbaric oxygen are beyond the scope of this book and are reviewed by Liebelt (109). The biological effects of carbon monoxide on pregnant mothers, fetuses, and newborn infants are reviewed by Ginsberg and Myers (110) and Longo (111).
Alcohol
Fetal Alcohol Syndrome
Alcohol is believed to affect the immature nervous system through chronic in utero exposure or through acute maternal intoxication.
In 1851, over 2000 years after Plutarch noted “one drunkard begets another” and Aristotle observed that “drunken women bring forth children like to themselves,” Carpenter reviewed the then current data on alcohol use during pregnancy and concluded that offspring can experience mental and behavioral disabilities (112). More than 100 years later, French (113,114) and American (115) clinicians described a craniofacial and neurologic disorder with growth and psychomotor retardation called the fetal alcohol syndrome (FAS). Its frequency, as estimated by Clarren and Smith in 1978, is between 1 and 2 in 1,000 live births, with partial expression of the syndrome, the fetal alcohol effects syndrome (FES), occurring in 3 to 5 in 1,000 live births (115). A survey published in 1997 indicated that the combined rate of FAS and FES was at least 9.1 in 1,000 live births (116).
Data obtained from a variety of experimental models suggest that alcohol, or a metabolic product thereof, such as acetaldehyde, can adversely affect the developing brain through multiple mechanisms. These include direct effects such as a transient impairment in uterine blood flow, vasoconstriction of uterine vessels, and reduced fetal cerebral metabolic rate. In addition, alcohol, which acts as an N-methyl-D-aspartate (NMDA) antagonist and has GABA-mimetic properties, is capable of triggering apoptotic neurodegeneration (117). The various potential mechanisms by which alcohol can damage the developing nervous system have been reviewed by West and colleagues (118), Shibley and Pennington (119), and Davies (120).
Despite the wealth of experimental data, considerable debate surrounds the pathogenesis of FAS and FES. On the one hand, some researchers question whether there is a threshold amount of alcohol intake, beyond which fetal abnormalities can develop, and suggest that even the smallest amount of alcohol can be dangerous to the fetus. On the other hand, it is not known what role several other variables play in the production of FAS and FES. These variables include the type and quality of the alcoholic beverage consumed (particularly the presence of contaminants in illicitly procured alcohol), associated food and vitamin deficiencies, binge drinking, smoking during pregnancy, drug intake, low plasma zinc levels, and the stage of pregnancy when exposure occurred (121,122,123). Furthermore, we do not know whether there are certain alleles of alcohol dehydrogenase that are protective or, conversely, place the fetus at increased risk for FAS (124). Also not established is whether the same dysmorphology can occur in the absence of maternal alcohol intake. Lacking such data, it is unclear whether alcohol per se causes FAS and FES, or whether high maternal alcohol intake is but a reflection of other environmental or genetic causes of mental retardation.
Neuropathologic features of FAS include neuroglial and leptomeningeal heterotopias with aberrant neural and glial tissue incorporated within arachnoid and meninges (125). Lissencephaly, cerebellar dysplasia, and agenesis of the corpus callosum also have been described, and migration anomalies can involve the cerebellum and brainstem as well as the cerebrum (126). MRI studies have delineated the variety of anomalies that can be encountered in FAS (127,128). Using material obtained for the most part during voluntary medical abortions, Konovalov and colleagues have shown that the proportion of fetuses showing these abnormalities in brain development increases with an increase in maternal ethanol intake (129), being lowest in women who were binge drinking during the first 3 months of pregnancy. They suggest that the teratogenic period for induction of the brain defects involves the initial 3 to 6 weeks of brain morphogenesis. These observations are at variance with those of Autti-Rämö and colleagues, who found that the number of physical anomalies was significantly increased in infants exposed to alcohol throughout gestation, but not when heavy alcohol consumption, defined as in excess of 140 g per week, was limited to the first or to the first and second trimesters (130).
The diagnosis of FAS is based entirely on history and the physical appearance of the child. In addition to confirmation of maternal alcohol abuse, the diagnostic criteria recommended by the Research Society on Alcoholism Fetal Alcohol Study Group require the following three: (a) the presence of prenatal or postnatal growth retardation; (b) CNS dysfunction such as developmental delay or
intellectual impairment; and (c) at least two of the following: microcephaly, microphthalmia, short palpebral fissures, or all three, and hypoplastic philtrum with thin upper lip and flattening of the maxillary area (131) (Fig. 10.1A,B). When a child demonstrates two of the three criteria, the condition is diagnosed with fetal alcohol effect (FAE). The use of this term has been challenged by Aase and colleagues on the basis of its imprecision, and that by labeling a child as having FAE, health workers presuppose that maternal alcohol abuse is the major or only cause of the child’s problems (132).
intellectual impairment; and (c) at least two of the following: microcephaly, microphthalmia, short palpebral fissures, or all three, and hypoplastic philtrum with thin upper lip and flattening of the maxillary area (131) (Fig. 10.1A,B). When a child demonstrates two of the three criteria, the condition is diagnosed with fetal alcohol effect (FAE). The use of this term has been challenged by Aase and colleagues on the basis of its imprecision, and that by labeling a child as having FAE, health workers presuppose that maternal alcohol abuse is the major or only cause of the child’s problems (132).
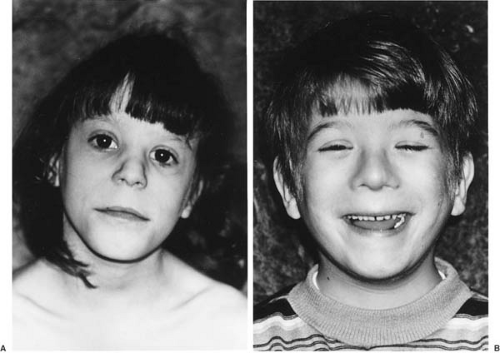 FIGURE 10.1. A: Fetal alcohol effects. This eight-year-old girl is mildly retarded and growth-deficient, with facial features typical of fetal alcohol syndrome: short palpebral fissures, short nose with flat nasal bridge, and thin lips with a smooth philtrum. B: This eight-year-old boy shows similar facial features. He is mildly retarded and growth-deficient. He weighed 2 kg at birth after a term gestation in a gravida 5, para 4, abortus 1, 28-year-old woman who drank one-fifth of hard liquor every one to two days during the first four months of pregnancy. (Courtesy of Dr. John Graham, Cedars-Sinai Medical Center.) |
Optic nerve hypoplasia is seen in up to one-half of children born to alcoholic mothers (133). Other ocular anomalies include microphthalmia, glaucoma, and cataracts (134). Less often, a sensorineural hearing loss and a variety of other minor physical anomalies occur (130). Outside the nervous system, abnormalities of the heart (notably ventricular septal defects), joints (e.g., congenital dislocation of the hips and flexion-extension deformities), genitalia, or skeleton occur (135,136).
A considerable amount of work has been done to ascertain the long-term cognitive development of children diagnosed in infancy as having FAS or FAE. Two such studies can be cited as examples of the problems encountered in this endeavor. In the Finnish study of Autti-Rämö and colleagues, children who were subjected to heavy alcohol exposure throughout gestation had significantly lower scores on the Bayley Mental Scale and the Reynell Verbal Comprehension Test at 27 months of age. This study, however, failed to find any significant effect on intellectual development when heavy prenatal alcohol exposure was restricted to the first trimester. In this study, as in many others, social class and maternal education could have been confounding variables (137). In the German study of Spohr and coworkers, children were reexamined more than 10 years after their initial diagnosis (138). Many of the
features of FAS and some of the minor physical anomalies had disappeared in the interval, so it became difficult to diagnose FAS in a substantial proportion of children who initially had a mild expression of FAS. In severely affected children with FAS, the features of the condition tended to persist. In this series (138), 88% of FAS children were microcephalic at the first assessment at 3 years of age and 65% at the second assessment at approximately 11 years of age. There was little improvement in IQ scores, and 69% of children had an IQ of 85 or less. It was noteworthy that not all children with mild morphologic damage had normal intelligence, and not all children with severe morphologic changes were mentally retarded. These findings are at variance with the observations of Mattsen and colleagues (139), who found that alcohol-exposed children displayed significant deficits in overall IQ measures even in the absence of any dysmorphic features.
features of FAS and some of the minor physical anomalies had disappeared in the interval, so it became difficult to diagnose FAS in a substantial proportion of children who initially had a mild expression of FAS. In severely affected children with FAS, the features of the condition tended to persist. In this series (138), 88% of FAS children were microcephalic at the first assessment at 3 years of age and 65% at the second assessment at approximately 11 years of age. There was little improvement in IQ scores, and 69% of children had an IQ of 85 or less. It was noteworthy that not all children with mild morphologic damage had normal intelligence, and not all children with severe morphologic changes were mentally retarded. These findings are at variance with the observations of Mattsen and colleagues (139), who found that alcohol-exposed children displayed significant deficits in overall IQ measures even in the absence of any dysmorphic features.
Several other studies purport to demonstrate that even children whose IQs fall within the normal range have deficits in cognitive function, particularly in language development, and that school performance tends to deteriorate with time (138,140,141). It is quite likely that an adverse social environment contributes substantially to these features.
Acute Alcohol Poisoning
Acute alcohol intake by infants and young children initially induces a depression of the CNS, and if untreated, it causes marked hypoglycemia and convulsions several hours after ingestion (142,143,144). The mechanism for hypoglycemia is not completely understood. Wilson and coworkers suggest that ethanol inhibits hepatic gluconeogenesis because of an increase in the ratio of nicotinamide-adenine dinucleotide (reduced form) (NADH) to nicotinamide-adenine dinucleotide (NAD), and possibly also because it inhibits the carboxylation of pyruvate to oxaloacetate (144).
Theophylline (Aminophylline)
Theophylline, like other alkylxanthines, lowers the seizure threshold by antagonizing the anticonvulsant effect of endogenous adenosine (145). Neurological symptoms can result from administration of a toxic dose of the drug, or from a therapeutic dose in children who have a lowered seizure threshold as a result of a preexisting seizure disorder or other previous brain injury or disease (146). Seizures are usually the most serious complication. These are often accompanied by cerebral vasoconstriction and ischemia. Ataxia, visual hallucinations, dyskinesias, and infantile spasms also have been reported as complications of theophylline toxicity (147,148,149). In premature infants, toxic effects include tachycardia, jitteriness, clonic posturing, and generalized seizures (150). Treatment of seizures involves the various anticonvulsants used for status epilepticus (see Chapter 14). Exchange transfusions also have been used in neonates given toxic dosages of theophylline.
Barbiturates
With the increased availability of other sedatives, the incidence of barbiturate intoxication in infants and children has diminished substantially since the 1980s.
Pharmacology
Barbiturates are general depressants. The mechanism of their action has been reviewed by Olsen and coworkers (151). In essence, toxic effects on the central nervous system (CNS) result from the particular sensitivity of the reticular activating system to barbiturates. On a subcellular basis, barbiturates act on the γ-aminobutyric acid (GABAA) receptor sites of postsynaptic neurons to increase the GABA-induced chloride current, thus potentiating the inhibitory effects of GABA. In addition, they inhibit excitatory α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors.
The severity of intoxication induced by a given dose of barbiturate varies considerably. At comparable blood levels, slow-acting barbiturates are less toxic than rapid-acting thiobarbiturates. Other factors affecting the severity of clinical symptoms are the development of tolerance, as occurs in children treated with barbiturates for a convulsive disorder, and the rapidity with which the drug is metabolized by the liver.
Clinical Manifestations
Clinical symptoms of barbiturate intoxication are mainly those of CNS depression. A modified Glasgow Coma Scale, which scores both the level of consciousness and vital signs (respiratory rate, blood pressure, and hypothermia), has been suggested as a means of grading the severity of barbiturate intoxication (152). In severe intoxication, the child is comatose, although the deep tendon reflexes can persist and the plantar reflex is often positive. Barbiturates are respiratory depressants; they impair respiratory drive and the mechanisms responsible for rhythmic respiratory movements. Respiratory centers become insensitive to increased CO2, and respiratory control becomes dependent on hypoxic drive, which, in turn, fails with progressively increasing barbiturate levels. As a consequence, respiration is affected early, and breathing becomes slow and shallow. Blood pressure decreases as a direct action of the drug on the myocardium and the depression of the medullary centers. A central antidiuretic action depresses urine formation. Hypoxia and pulmonary complications are common with severe barbiturate poisoning. Neuropathologic findings closely resemble those of fatal anoxia.
Diagnosis
The diagnosis of barbiturate intoxication in a child with severe CNS depression or coma rests on the history of drug ingestion or, in the absence of such a history, on the exclusion of other causes for coma and on the positive identification of the toxin.
The physical examination, which must be performed expeditiously, should be directed toward the exclusion of infectious, traumatic, metabolic, and vascular causes for the child’s condition. Nuchal rigidity and focal neurologic signs make drug ingestion unlikely. Mitchell and colleagues (153) have stressed the frequency of miosis in children who are comatose owing to barbiturate intoxication and noted that miosis is usually absent in children admitted in coma as a consequence of head trauma or infection. The physician must always consider the possibility of nonaccidental barbiturate ingestion, which is an increasing form of child abuse (154).
A number of rapid urinary screening tests are available that produce characteristic color reactions in the presence of barbiturates or other toxins.
Treatment
Because barbiturate levels in brain correlate poorly with those in serum, serum barbiturate levels can only serve as a guide to assess the severity of intoxication. If the drug was ingested shortly before the child is examined, gastric lavage is done. When respirations are depressed, an airway is instituted, and artificial respiration is used whenever necessary to maintain normal blood CO2 and pH levels. Circulatory collapse, the chief cause of death in barbiturate intoxication, is prevented with plasma expanders or whole blood. Vasopressors are of relatively little value except in the initial management of severe hypotension (155). Forced diuresis with fluid loading and alkalinization by the intravenous administration of sodium bicarbonate facilitate urinary excretion of phenobarbital and other long-lasting barbiturates. Renal failure and hypothermia are other potentially serious complications of barbiturate intoxication (156).
In severe intoxication, or in children who because of renal or hepatic disease have impaired drug elimination, hemodialysis or peritoneal dialysis is the most effective means of removing the drug from the bloodstream (157). The effectiveness of peritoneal dialysis depends on the blood barbiturate level, which is usually high when long-acting barbiturates are ingested. Multiple doses of charcoal appear to be of little benefit.
Detoxification with hemoperfusion through charcoal coated with acrylic hydrocarbons and exchange transfusion also has been suggested (158). The use of analeptics such as picrotoxin has been abandoned because they were found to induce cardiac arrhythmias and convulsions.
The presentation of benzodiazepine overdose is similar to that of barbiturate intoxication, and treatment is analogous, except that diuresis and forced alkalinization are unnecessary, because diazepines are metabolized by the liver. As a class, benzodiazepines are safer than barbiturates, and the mortality for intoxications is far smaller (156).
Phenothiazines
The neurotoxic actions of phenothiazines result from either an overdose or idiosyncratic adverse reactions to therapeutic dosages. The toxic responses include depression of consciousness, seizures, hypotension, loss of temperature control, and movement disorders. In the series of James and coworkers, 90% of children who accidentally ingested phenothiazines or other psychotropic medications presented with depressed consciousness, while 51% developed dystonia (159). Movement disorders constitute the major adverse effects of therapeutic doses of phenothiazines. Although all phenothiazines can produce movement disorders, the extrapyramidal signs in children occur most commonly with prochlorperazine, haloperidol, thioridazine, chlorpromazine, and clozapine (160). They are less likely to occur with olanzapine and risperidone.
Several kinds of movement disorders have been encountered. Younger children tend to have generalized manifestations, whereas adolescents tend to have signs localized to the face and trunk (160). The most common are sudden episodes of opisthotonus accompanied by marked deviation of the eyes and torticollis without loss of consciousness. Dystonic movements of the tongue, face, and neck muscles, drooling, trismus, ataxia, tremor, episodic rigidity, and oculogyric crises also are seen. Seizures are rare but can be mimicked by violent dystonic movements.
Tardive dyskinesia is a late appearing neurological syndrome seen with the use of phenothiazines and other neuroleptic drugs, and is believed to be the consequence of hypersensitivity of dopamine receptors in the basal ganglia secondary to prolonged blockade of the receptors by the medication. The condition is relatively uncommon in children, and there is less risk with olanzepine and the other atypical antipsychotic agents. Tardive dyskinesia is characterized by stereotypic choreiform movements of the face, eyelids, mouth, and tongue and less so of the trunk and extremities. Dystonic movements also can be seen, as can a tardive akathisia, an unpleasant sensation of internal restlessness that is relieved by volutional movements.
The condition persists even following withdrawal of the offending medication. In the Queen Square series of Kiriakakis, remission occurred after a mean of 5.2 years following onset (161). A large number of agents have been used to treat this condition, notably tocopherol, propanolol, lithium, and clonidine, but none of these appears to have a convincing benefit. Most recently,
branched-chain amino acids have been suggested for the treatment of tardive dyskinesia in children and adolescents (162). Tardive dyskinesia is also discussed in Chapter 3.
branched-chain amino acids have been suggested for the treatment of tardive dyskinesia in children and adolescents (162). Tardive dyskinesia is also discussed in Chapter 3.
A neuroleptic malignant syndrome has occasionally been encountered. The most common inciting drugs are haloperidol, phenothiazines, butyrophenones, and thioxanthines. The condition generally develops 3 to 9 days after initiation of neuroleptic treatment. It manifests by hyperthermia, muscular rigidity, extrapyramidal signs, opisthotonus, altered consciousness, and autonomic disturbances. Hyperthermia does not always have to be present (163). Serum creatine phosphokinase is elevated, and myoglobinuria is common. The condition lasts approximately 7 to 10 days (164). In contrast to its frequency in patients who have ingested toxic doses of phenothiazines, drowsiness is rare when symptoms are caused by an idiosyncratic reaction to the drug. Persistence of the dyskinesias after withdrawal of the drug is relatively common in young adults, but rare in children (165).
Extrapyramidal movements have been reported in newborns whose mothers received phenothiazines during pregnancy. The movement disorder is accompanied by hypertonia, tremor, and a poor suck reflex. It can persist for up to 12 months (166).
Diagnosis of phenothiazine intoxication depends on the history of drug ingestion. The most common appearance of the extrapyramidal disorder, namely as episodes of opisthotonus, trismus, and dystonic posturing, is distinctive. Paroxysmal choreoathetosis can present a similar picture, but its onset is gradual with a prolonged history of recurrent attacks.
Diphenylhydramine in a dose of 2 mg/kg, given intravenously over the course of 5 minutes, is an effective antidote for the extrapyramidal disorder appearing at therapeutic doses, and it often produces a dramatic response. Benztropine mesylate (0.5 mg/kg), given intravenously, or diazepam also is effective (160).
Neuroleptic malignant syndrome is treated by discontinuation of the neuroleptic, reduction of hyperthermia by means of cooling blankets, and support of respiratory and cardiovascular status. The administration of dantrolene (0.5 mg/kg every 12 hours) or bromocriptine has been suggested, but controlled studies do not appear to show that either drug administered singly or in combination shortens the illness (167).
Antibacterial Agents
The increasing number of antibacterial agents used by clinicians for the treatment of various infectious diseases also has increased the incidence of neurologic complications associated with antibacterial therapy. The various clinical syndromes of neurotoxicity are shown in Table 10.4. They also have been reviewed by Thomas (168) and Wallace (169).
TABLE 10.4 Neurotoxicity of Antibacterial Therapy | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
Encephalopathy/Seizures
The β-lactam antibiotics, notably penicillins, cephalosporins, and carbapenems, can manifest significant neurotoxicity. Studies on animal models have shown the toxic effect of penicillins to be caused by a dose-dependent inhibition of the GABA-gated chloride ion influx (168,170).
The principal manifestations of β-lactam toxicity are seizures. The major risk factors for development of seizures are high-dose intravenous use of the antibiotic, impaired renal function, a preexisting or concurrent CNS disorder such as meningitis, and the concomitant use of drugs such as theophylline, which can lower the seizure threshold (169). In addition to seizures, the β-lactams can cause hallucinations, and in the case of ceftazidime, absence status epilepticus (169).
Cochlear and Vestibular Damage
Several aminoglycoside antibiotics can induce damage to the eighth nerve. Gentamicin and tobramycin can damage both cochlear and vestibular components, whereas neomycin, kanamycin, and amikacin affect the auditory component predominantly. These complications are less common in children than in adults. Routine studies of brainstem auditory-evoked potentials during aminoglycoside therapy can detect high-frequency hearing loss as an early sign of ototoxicity (171). The development of ototoxicity is related to the dosage and duration of aminoglycoside therapy, the presence of impaired renal function, and a genetic predisposition to aminoglycoside susceptibility, notably a mitochondrial ribosomal RNA mutation (172). Symptomatic hearing loss is seen initially as reduced high-frequency acuity, followed by impairment of lower-frequency hearing, damage to the hair cells, and complete destruction of the organ of Corti with ultimate eighth nerve damage.
Neuromuscular Blockage
A myasthenic syndrome has been encountered in children treated with neomycin, ampicillin, gentamicin, and other aminoglycoside antibiotics. These drugs should therefore be used cautiously in children with neuromuscular disease, notably myasthenia gravis (173).
Pesticides
Organophosphates
Organophosphate insecticides are widely used on food crops throughout the world. These chemicals inhibit acetylcholinesterase and result in an accumulation of acetylcholine and overstimulation of the muscarinic postganglionic fibers of the parasympathetic nervous system. When ingested or inhaled, manifestations are of three types: acute, intermediate, and delayed. The acute syndrome represents a cholinergic crisis with headache, abdominal pain, irritability, sweating, miosis, and muscular twitching (174). After recovery, which generally occurs within 24 to 96 hours of the poisoning, an intermediate syndrome develops in many patients. This is characterized by profound muscular weakness, affecting predominantly the proximal and respiratory muscles, and often accompanied by cranial nerve palsies. Dystonia was seen in approximately 20% of Sri Lanka patients (175). Symptoms resolve within 1 to 5 weeks. A delayed polyneuropathy appears 2 to 3 weeks after the poisoning and often blends into the intermediate phase (176). It primarily affects the distal musculature, with recovery in 6 to 12 months. Controversy exists as to whether neurobehavioral effects persist after recovery (177).
Atropine has been recommended for treatment during the acute phase. For children younger than 12 years of age, the dose is 0.05 mg/kg, given intravenously. This is followed by a maintenance dosage of 0.02 to 0.05 mg/kg every 10 to 30 minutes until the cholinergic signs are no longer present or until pupils become dilated. Pralidoxime, given intravenously over the course of 15 to 30 minutes in a dose of 25 to 50 mg/kg, also has been recommended (178).
Organochlorines
Endrin is the most toxic of the organochlorine pesticides and has been responsible for several epidemics throughout the world. Symptoms are caused by CNS excitation. This results from suppression of the GABA-induced chloride current, and with it, suppression of the GABA-mediated synaptic inhibition (179). Symptoms occur within 12 hours of the poisoning. Convulsions are frequently the presenting symptom. Older children report headache, nausea, and muscle spasms just before the seizures (180). Phenobarbital or benzodiazepines are generally effective in controlling seizures, although recurrent convulsions are not unusual (181).
Lindane, another chlorinated hydrocarbon, has been used extensively in the form of Kwell for the treatment of scabies, and its accidental ingestion by toddlers is not unusual. If ingested in sufficient amounts, the substance can induce depression of consciousness, coma, and major motor seizures (182). The clinical picture of exposure to the various types of pesticides is reviewed by O’Malley (177).
Psychedelic Drugs
Fashions occur in psychedelic drugs, as they do in clothes. Currently, the use of 3,4-methylenedioxy-methamphetamine, also known as ecstasy, among college students and adolescents continues to exceed that of lysergic acid diethylamine and cocaine (183). A recently published American survey indicated that 5% of tenth graders had used ecstasy within the last year, and 2% reporting using it within the last month (184). Data obtained from New Zealand show a comparable incidence. Accidental poisoning with psychedelic drugs is uncommon in small children and is generally not life threatening, although subarachnoid hemorrhage following use of the drug has been reported (185).
Intoxication with phencyclidine often is produced by passive inhalation (186,187). The psychosis seen in older subjects is less common in small children than a depressed sensorium, hypotonia, dystonia or opisthotonus, and ataxia. Seizures as well as opisthotonus and a variety of ocular signs, notably horizontal or vertical nystagmus and miosis, can be encountered after phencyclidine or ecstasy ingestion (186,188). Treatment is symptomatic, and observation of the intoxicated youngster usually suffices. A variety of barbiturates or phenothiazines can be used for sedation.
Hexachlorophene
Hexachlorophene—a chlorinated phenolic compound widely used as an antiseptic until 1972, when it became a prescription drug—has been shown to induce seizures when absorbed through burned or extensively excoriated skin. Significant amounts also are absorbed through the intact skin of premature neonates (189).
Accidental ingestion of hexachlorophene by older children results in vomiting, hypotension, and a neurologic picture characterized by total blindness caused by deterioration of the optic nerves (190). Additionally, behavioral changes, convulsions, increased intracranial pressure, paralysis, and spastic paraparesis or paraplegia can occur (191).
Mepivacaine
In the past, the use of mepivacaine hydrochloride to induce paracervical or pudendal block in the course of labor resulted in accidental poisoning of the newborn by direct injection of the anesthetic. The clinical picture was reduced responsiveness, bradycardia, apnea, hypotonia, and seizures that commenced within 6 hours, usually between 1 and 3 hours of age. Mepivacaine intoxication is differentiated from perinatal asphyxia by the earlier onset of seizures and the abnormality of oculomotor reflexes, which in asphyxia tend to be preserved for the first few hours of life. The condition is self-limiting, and if asphyxia is prevented, infants return to normal by 18 hours of age (192).
Salicylate
A less widespread use of salicylates in this country has resulted in a significant reduction in the incidence of salicylate intoxication. Currently, most acute intoxications are the outcome of attempted suicide with aspirin or methylsalicylate (193). Both acute and chronic salicylate intoxication are associated with mental status depression whose severity is usually proportionate to the serum salicylate level (194). Additionally, a delayed encephalopathy owing to cerebral edema can develop. In some instances, this encephalopathy is associated with a rapid decrease in serum sodium concentrations, the consequence of inappropriate antidiuretic hormone secretion resulting from the action of salicylates on the brain (195). The administration of salicylates in a patient with an influenza virus or varicella infection can initiate Reye syndrome (see Chapter 7).
Eosinophilia-Myalgia Syndrome
Eosinophilia-myalgia syndrome is characterized by eosinophilia, myalgia, and a progressive neuropathy. Muscle biopsy demonstrates a perivascular eosinophilic infiltrate; sural nerve biopsy documents a demyelinating neuropathy with axonal degeneration (196). Involvement of the CNS includes seizures, dementia, and vascular infarctions (197). The condition is induced by consumption of tryptophan contaminated with 3-(phenylamino) alanine. This substance is chemically similar to 3-phenylamino-1,2-propanediol, an aniline derivative believed to be responsible for the contamination of rapeseed oil, which caused the toxic oil syndrome seen during 1981 in the central region of Spain (198). Cooking oil contaminated with polychlorinated biphenyls has been responsible for mass poisoning in Japan and central Taiwan. Neurologic symptoms include polyneuropathy. Children who were in utero when their mothers consumed contaminated oil were prone to severe developmental deficits (199).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


