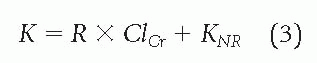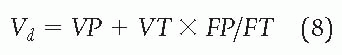Management of seizures in the presence of renal and liver disease has become an increasingly common problem during the past several decades. Prolonged survival, achieved largely through advances in dialysis, pharmacology, and transplantation, accounts for a growing population of patients with altered metabolic capacities. The emergence of opportunistic hepatic infections in acquired immune deficiency syndrome and other immunocompromised conditions, as well as the prevalence of viral hepatitis, has increased the population with impaired liver function. Pharmacologically induced renal dysfunction and systemic diseases, such as hypertension and diabetes, continue to occur frequently in patients whom an epileptologist may encounter. Consequently, neurologists must possess a basic understanding of pharmacology and the specific pharmacokinetics of anticonvulsants in liver and renal disease.
Patients with pre-existing liver and renal disease may require transient treatment with anticonvulsants for seizures as a result of electrolyte shifts associated with worsening uremia and dialysis, as well as hepatic insufficiency caused by chronic alcohol abuse. Secondary effects of disease in either of these organs can adversely affect blood pressure and coagulation, resulting in potentially epileptogenic cerebrovascular events. In addition, patients with epilepsy are not immune to the liver and renal diseases that occur in the general population. Antiepileptic drugs (AEDs) themselves may induce such organ dysfunction, complicating or contraindicating their further use.
This chapter reviews the clinical use in liver and renal disease of the commonly prescribed AEDs and the newer anticonvulsant medications that became available between 1993 and 2004. Because the degree of debility and the response to AEDs vary significantly among patients, specific rules cannot be inferred, and practical guidelines only are offered. The general biopharmacologic principles that precede the discussion of specific agents apply not only to current anticonvulsant therapy but also to drugs potentially available in the future.
MEDICATIONS IN RENAL DISEASE: OVERVIEW
The degree to which renal disease alters the pharmacokinetics of specific drugs depends on their primary mode of elimination. Drugs excreted unchanged by the kidneys have a slower rate of elimination and longer half-life in patients with renal disease than in healthy persons, increasing drug accumulation and necessitating lower doses and longer interdose intervals to prevent toxic effects.
Drugs are divided into three classes: (a) type A, which are eliminated completely by renal excretion; (b) type B, which are eliminated by nonrenal routes; and (c) type C, which are eliminated by both renal and nonrenal routes (
1,
2). Because the relationship between half-life and creatinine clearance (Cl
Cr) is not linear, dosing predictions based on renal insufficiency are difficult. However, estimates may be determined from the following linear equation that describes the speed of drug elimination as a function of creatinine clearance:
where
K is the elimination rate constant,
R the slope of
K against
ClCr, and
KNR is the rate constant for drug elimination by nonrenal routes (
4).
Nomograms based on computed values of K and KNR will predict new maintenance doses that are reduced proportionately to the reduction in K. However, such linear equations do not take into account the effect of renal insufficiency on drug biotransformation, elimination of metabolites with toxic properties, or decreases in plasma protein binding.
Studies show that some drug oxidations in liver endoplasmic reticulum can be accelerated in uremia (
5,
6). The mechanism is undefined, but several possibilities have been proposed. Poorly excreted nutritional substances that can induce microsomal drug metabolism may be present in excess quantities in renal patients. Indole-containing cruciferous plants (cabbage, cauliflower, Brussels sprouts) induce these enzymes in rats (
7). Drugs with low hepatic extraction and high protein binding will have higher rates of metabolism in uremia as the free fraction increases, which, in turn, increases plasma clearance and the apparent volume of distribution (
Vd):
where
VP is the plasma volume,
VT is the extravascular volume,
FP is the fraction of free drug in plasma, and
FT is the fraction of free drug in tissue (
9).
Protein binding of anionic acidic drugs (such as phenytoin, which is strongly bound by albumin) decreases in patients with renal dysfunction (
Table 50.1). Drugs with organic bases have variable protein binding in renal disease, and those that bind primarily to one site have decreased binding, an effect described in the literature since 1938 (
10). However, this reduced binding exceeds the amount that can be accounted for by a simple decrease in serum albumin. Two hypotheses relating to uremia have been proposed to resolve this discrepancy: the existence of small molecules that competitively displace drugs from normal binding sites (
11) and altered binding sites of albumin molecules (
12). Experimental evidence supports both mechanisms, and each probably is involved to some extent in individual patients (
13). Although drug metabolism may be accelerated in uremic humans, the same drugs may exhibit slowed metabolism in uremic animals, complicating the extrapolation from experimental data (
14).
Although dialysis ameliorates renal insufficiency, it alters the response to medications. The removal of drugs from serum by hemodialysis depends on numerous variables, including molecular weight, protein binding, plasma concentration, blood flow, and hematocrit, as well as on the inherent clearance characteristics of the dialyzer. Dialysis also can profoundly affect drug activity through changes in pH level, protein concentration, osmolality, electrolytes, and glucose and urea levels. Peritoneal dialysis, unlike hemodialysis, is influenced by vascular disease because the blood supply presented to the dialysate passes through arterioles. Drug additives are used more frequently in peritoneal dialysis than in hemodialysis solutions, creating a small potential for drug interactions (
15). Following hemodialysis, albumin binding of such medications as phenytoin and phenobarbital is decreased, perhaps because of increased levels of nonesterified fatty
acids, which bind strongly to albumin (
16). This effect has been proposed for heparin, administered systemically during dialysis, with resultant activation of lipoprotein lipase (
17).
MEDICATIONS IN LIVER DISEASE: OVERVIEW
Because the liver is a primary site of drug metabolism in humans, hepatic insufficiency can significantly alter biotransformation and disposition, although pathophysiologic changes will vary according to the disease or its stages. Hepatic blood inflow by the portal vein, hepatocellular mass, and functional capacity primarily determine the effects of liver disease on drug handling.
Table 50.2 illustrates the major changes found in cirrhosis, acute viral hepatitis, and alcoholic hepatitis (
18).
At least five categories of liver disease affect drug disposition: (a) chronic liver disease; (b) acute hepatitis; (c) drug-induced hepatotoxicity; (d) cholestasis; and (e) hepatic infiltrative/neoplastic disease. In addition, medications must be classified not only by protein binding but also by the capacity of the liver to extract drug as blood flows through the organ: flow limited, capacity limited with high protein binding, and capacity limited with low protein binding (
19).
Flow-limited drugs have high extraction rates, and clearance is limited primarily by blood flow. Their rate of metabolism depends on the amount of drug presented to the liver, which is proportional to blood flow. Most anticonvulsants are capacity-limited drugs, as their extraction ratios are low (<0.2).
Table 50.3 lists the extraction ratios of major antiepileptic compounds (
18,
20,
21,
22,
23). The rate of metabolism of capacity-limited drugs depends on the concentration of free drug at hepatic enzyme receptor sites and thus on the extent of protein binding. Capacity-limited, binding-sensitive drugs, such as phenytoin, valproic acid, and carbamazepine, are greater than 85% bound to plasma proteins at therapeutic concentrations; therefore, alterations in plasma protein concentration and binding characteristics can significantly alter their hepatic clearance (
23,
24). Capacity-limited, binding-insensitive drugs, such as ethosuximide, have a low affinity for plasma protein (usually less than 30% at therapeutic concentrations), and clearance is only minimally affected by changes in protein binding.
The following model, combining the principles of intrinsic metabolic capacity and blood flow, has been proposed:
where
Clh is the volume of blood cleared by the liver per unit time,
Q is total hepatic blood flow,
Fb is the fraction of drug bound to protein and cells, and
Clint is the intrinsic metabolic clearance (
23,
24,
26).
The latter term, defined as the volume of liver water cleared of drug per unit time, varies directly with the Michaelis constant. The extraction ratio (E) may be derived by dividing hepatic blood flow into total hepatic clearance:
When combined hepatic and renal clearance occurs, clearances are additive.
Although hypoalbuminemia is frequently a feature of liver disease, drug binding to plasma proteins may be decreased even without measurable changes in albumin concentration (
Table 50.4). Mechanisms similar to those causing decreased protein binding in renal insufficiency have been suggested (
28,
29). Because intrinsic clearance varies with the type and duration of liver disease, the effects of changes in protein binding in capacity-limited, binding-sensitive drugs are complex. If hepatic disease lowers binding without changing intrinsic clearance, total drug concentration will ultimately fall because the rate of metabolism of these drugs depends on the free fraction. If liver disease reduces intrinsic clearance, total drug concentration may remain the same or increase as the free concentration increases. This can result in enhanced response or toxic effects at lower than expected drug levels and may explain the increased incidence of adverse reactions to medications such as valproic acid in liver disease (
30).
Capacity-limited, binding-insensitive drugs can be considered relatively pure indicators of intrinsic clearance. However, tissue binding to substances such as ligandin may contribute to the volume of distribution of drugs and thus to their half-life. The effect of liver disease on the content and function of such binding proteins is poorly understood. Tissue binding can be affected by secondary pathologic changes of liver disease, such as alterations in tissue and plasma pH level (
24,
31,
32,
33) or by ascites (
34,
35).
Because of various types of drugs and stages of liver disease, as well as interindividual variation, predicting changes in drug kinetics in patients with hepatic insufficiency remains difficult. Studies have identified additional discrepancies between observed changes and those suggested by pharmacokinetic predictions (
12,
23,
26). Another variable is the potential for autoinduction of microsomal enzymes after long-term drug administration. Phenobarbital and carbamazepine and their active metabolites have this potential, with resultant temporal variability in drug levels and efficacy, increased complexity of drug interactions, and the potential increased risk of liver dysfunction.