Tumors of the Nervous System
Bernard L. Maria
John H. Menkes
BRAIN TUMORS
Incidence
Tumors of the central nervous system (CNS) occur relatively frequently during the early years of life. They are the most common solid tumors of childhood and afflict approximately 1,500 patients every year in the United States (1,2). Reported incidence rates have varied from 20 to 50 per 1,000,000 children (3). Representing 20% of all malignancies, they are second only to leukemia in overall cancer incidence and account for a high proportion of deaths (4). CNS tumors in infants less than one year old account for about 10% of all intracranial tumors occurring in the pediatric population (5). Males and white, non-Hispanics have a higher incidence of CNS tumors compared with females and other ethnic groups, respectively (6). The nadir in incidence of CNS neoplasms between the ages 0 and 44 years occurs in the 15- to 29-year age group, at a rate of 22.3 per million (5).
Although the incidence of leukemia has remained relatively stable, the incidence of brain tumors has increased approximately 2% per year in the past 20 years (7). A review of earlier studies on the incidence of intracranial space-occupying lesions indicates that the relative frequencies of pathologic varieties have changed. Critchley, writing in 1925, found tuberculoma to be the most common intracranial tumor of childhood (8). Subsequent surveys by Bailey and associates (9) and Cuneo and Rand (10) stressed the preponderance of gliomas. Meningiomas and neurinomas were virtually absent, except in association with neurofibromatosis (11). Tuberculomas are now rare in the developed countries, but a series of 107 cases was reported from India as recently as 1965 (12). Data from the National Cancer Institute Surveillance, Epidemiology, and End-Results (SEER) from 1975 to 1998 showed that the incidence of CNS tumors in people age 0 to 44 years increased over time, mainly due to an increased incidence of “other glioma” (5). From 1981 through 1993, the incidence of primitive neuroectodermal tumor/medulloblastoma (PNET/MB), the most common malignant posterior fossa tumor, rose more than 4% per year (Table 11.1). This trend has not been fully explained, but improved detection through sensitive and specific neuroimaging technology, such as magnetic resonance imaging (MRI), may account for much of the increase (13). Inherited conditions such as neurofibromatosis, tuberous sclerosis, basal cell nevus syndrome, Von Hippel–Lindau disease, Li-Fraumeni syndrome (p53 mutation), Gorlin syndrome (PATCHED mutation), and Turcot syndrome (APC mutation) account for a small percent of CNS tumors (14,15,16,17,18,19,20,21). In addition to inherited conditions, ionizing radiation delivered in the past for tinea capitis or for leukemia and brain tumors is a known risk factor for CNS tumors (22,23,24). Various risk factors including nitrosamine/micronutrient intake, pesticide/fertilizer exposure, and parental occupation in chemical industries have been inconsistently associated with childhood brain tumors (25). Thus, the etiology for the vast majority of CNS tumors is unknown and there is no conclusive data to show that toxic waste, air and water pollutants, and electromagnetic fields and irradiation in local environments are tumorigenic (26). Furthermore, environmental influences ordinarily are expected to be gender blind, and thus could not account for the 4% increase in PNET/MB, which elicits a male-to-female gender ratio of approximately 2.5:1. Epidemiological investigations and population-based case-control studies are needed to determine how genes, environment, and lifestyle influence risk and may combine to cause CNS tumors in children (27).
Pediatric brain tumors include a spectrum of both glial and nonglial tumors that differ significantly in location and biologic behavior from that of adults (Table 11.2) (28,29). Subtentorial tumors constitute more than 50% of all intracranial space-occupying lesions in children; in adults, only 25% to 30% of tumors originate below the tentorium. In children younger than age 1 year, as in adults, supratentorial tumors are most frequent; in infancy these often arise from hamartomas or other congenital malformations (1).
TABLE 11.1 Florida Childhood Cancer Incidence 1981–1993, 0–14 Years | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Pathogenesis
A discussion of the molecular origins of neoplasia within the nervous system is far beyond the scope of this text. However, in recent years, real progress has been made in our understanding of the molecular genetic abormalities that govern the initiation and/or progression of these tumors (30). Suffice it to say that the uncontrolled proliferation of a population of somatic cells is basically a genetic disorder, with multiple genetic abnormalities preceding the neoplastic transformation of normal tissue. Most tumors of childhood are derived from the neuroectoderm, and there is evidence that transcription factors expressed by neural crest cells correlate with phenotype in neurorectodermal tumors (31). Two distinct fundamental mechanisms appear to be operative. One is associated with the activation or overexpression of growth-promoting factors such as proto-oncogenes, and the other is the result of loss or inactivation of genes that normally regulate or suppress cell growth, the tumor-suppressor genes. The mutation of the proto-oncogene to an oncogene has a dominant effect; only one of the cell’s two gene copies needs to undergo the mutation. The inactivation of a tumor-suppressor gene is recessive, which means that both gene copies must be inactivated. Inactivation of tumor-suppressor genes is believed to be responsible for the development of tumors in neurofibromatosis and tuberous sclerosis (see Chapter 12).
TABLE 11.2 Incidence of Brain Tumors in the Pediatric Population Younger Than 15 Years of Age Versus All Agesa | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
The role of tumor viruses in CNS neoplasia of childhood is less clear. However, one-half of choroid plexus tumors and most ependymomas contain and express a segment of a gene related to the monkey polyoma virus (SV40). This virus, which is highly tumorigenic in rodents, could have an etiologic role in the development of some childhood neoplasms. The JC virus a human neurotropic polyomavirus, which causes progressive multifocal leukoencephalopathy and is closely related to SV40, has been detected in ependymomas and choroid plexus papilloma, but not medulloblastoma or pilocytic astrocytoma (32). It is also possible that certain human neoplasms provide a favorable microenvironment for viral replication in tissue that is latently infected with SV40 (33). From the data currently available, it is clear that the combination of accumulating events is distinct for each tumor type; what is still unknown is which molecular abnormality is the rate-limiting step that leads to neoplastic transformation.
Cytogenetic analysis of glial tumors has identified a variety of gross chromosomal abnormalities (Table 11.3) (34,35,36,37,38,39,40,41,42,43). Gains of chromosome 7 and losses of 9 and 10 have been reported in adult high-grade astrocytomas, but pediatric astrocytomas may also be associated with gains in chromosomes 1q and 2 (44). Other genomic imbalances include extra copies of chromosome 8q, loss of 6, 8, 10q, 11, 13, 17p, or 22, and structural abnormalities of chromosome 9p and 19q. Deletions of the short arm of chromosome 17 (17p13.3) is seen in approximately 40% to 50% of primary PNET/MB (30,45). Such deletions of 17p may occur in the absence of other gross abnormalities of 17, as a component of an isochromosome of 17q [i(17q)]. The molecular consequences of chromosomal loss in medulloblastoma are still being elucidated, but loss of 17p13.3, high-frequency MYC amplification, and Survivin expression are associated with a poor prognosis (45,46,47). Dysregulation of the hedgehop, PTCH, neurotrophin, ErbB receptor, adenomatous polyposis coli gene, and the Wnt signalling pathways that are important in CNS development has been shown in embryonal tumors, and current evidence favors site-specific origin of such tumors (48,49,50). ErbB2 is undectable in developing cerebellum but is expressed in 40% of medulloblastomas and is associated with a poor clinical outcome (51). The epigenetic silencing of tumor-suppressor gene expression by aberrant methylation (e.g., promoter hypermethylation) has emerged
recently as a major mechanism of tumorigenesis (52,53). P53 pathway dysfunction may also be abrogated in pediatric CNS tumors (54,54a). Moroever, polymorphisms among the genes involved in the folate pathway may significantly affect the risk for developing a childhood brain tumor (25). The molecular biology of tumors has been reviewed by Bigner and colleagues (34), Santarius and colleagues (55), and Ng and Lam (56). Complete and detailed reviews of newer developments in tumor cell culture, the effect of animal models on brain tumor research, and basic discoveries in neuro-oncology are also available elsewhere (57,58).
recently as a major mechanism of tumorigenesis (52,53). P53 pathway dysfunction may also be abrogated in pediatric CNS tumors (54,54a). Moroever, polymorphisms among the genes involved in the folate pathway may significantly affect the risk for developing a childhood brain tumor (25). The molecular biology of tumors has been reviewed by Bigner and colleagues (34), Santarius and colleagues (55), and Ng and Lam (56). Complete and detailed reviews of newer developments in tumor cell culture, the effect of animal models on brain tumor research, and basic discoveries in neuro-oncology are also available elsewhere (57,58).
TABLE 11.3 Chromosomal and Genetic Alterations Characteristic of Specific Types of Brain Tumors | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
From a clinical point of view, the importance of genetic factors in the development of brain tumors is significant. Surveys of the incidence of tumors in relatives of brain tumor patients have demonstrated a higher incidence of CNS neoplasms and of leukemia, but no greater incidence of other malignancies (59). Craniopharyngiomas and some of the PNET/MBs are congenital and arise from an area of maldevelopment. Craniopharyngiomas arise from persistent remnants of the craniopharyngeal (Rathke’s) pouch and PNET/MBs usually from primitive cell rests in the posterior medullary velum.
Postnatal factors, trauma in particular, have not proven significant in the development of brain tumors in children. In a few instances, irradiation has been implicated in the appearance of malignant mesodermal tumors at the radiation site. In one study, radiation for ringworm of the scalp was associated with an increased incidence of brain tumors occurring after an interval that ranged from 7 to 21 years (60).
Symptoms and Signs
Neurologic symptoms produced by brain tumors are general or local. General symptoms result from increased intracranial pressure, which results directly from progressive enlargement of the tumor within the limited volume of the cranial vault; local symptoms are due to the effects of the tumor on contiguous areas of the brain.
Pathophysiology
An intracranial mass produces cerebral compression by its intrinsic volume; by its encroachment on the ventricular system, obstructing the flow of cerebrospinal fluid (CSF) and producing ventricular dilatation; and particularly when malignant, by its ability to produce edema in white matter adjacent to the expanding mass.
As the tumor enlarges, the intracranial contents, primarily the ventricular spaces, are initially compressed. The duration of the initial asymptomatic period depends on the rate of tumor expansion and on whether the tumor’s location causes it to compromise the circulation of CSF.
When the intracranial pressure approaches or equals the systemic arterial pressure, it causes the systemic arterial pressure to increase. Cushing noted that systemic hypertension was accompanied by bradycardia and slow irregular respirations (Cushing triad) (61,62). Compression of the venous channels, especially the large, draining venous sinuses, reduces cerebral blood flow. The resulting cerebral anoxia in turn produces vascular dilatation and a further increase in intracerebral volume and pressure (63).
A number of experiments have proven Cushing’s contention that the pressure threshold for the induction of the vasopressor response is reached when intracranial pressure is at the level of systemic arterial pressure. Clinical experience, however, suggests that arterial hypertension first appears at lower pressures, particularly when a pressure differential exists above and below the tentorium. Under these instances, the pressure response can be triggered by local ischemia of the cerebral hemispheres or by distortion of the brainstem and subsequent medullary ischemia. With a further increase in intracranial pressure, the vasopressor mechanism fails, arterial pressure decreases, and cerebral blood flow is substantially reduced (64,65).
Pulmonary edema can accompany increased intracranial pressure (neurogenic pulmonary edema). Its pathogenesis is poorly understood; most likely, the edema is the direct result of an elevated pulmonary capillary pressure. Whether this elevation results from an increase in left atrial pressure (the consequence of left ventricular failure), as has been postulated for some years, or from a disturbance in hypothalamic function (with a consequent massive α-adrenergic discharge) has not been clarified (66). The rapid evolution of pulmonary edema after severe head trauma makes the latter alternative more attractive.
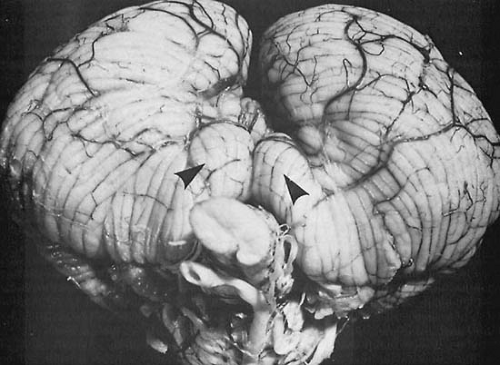 FIGURE 11.1. Herniation of cerebellar tonsils. The view from the posterior aspect of the cerebellum demonstrates caudal displacement of the cerebellar tonsils resulting from increased intracranial pressure. The cone of herniated tonsillar tissue (arrows) impinges on the dorsal surface of the cervical spinal cord. (From Bell WE, McCormick WF. Increased intracranial pressure in children, 2nd ed. Philadelphia: Saunders, 1978. With permission.) |
Increased intracranial pressure decreases cerebral blood flow when perfusion pressure (the difference between arterial pressure and intracranial pressure) decreases to approximately 40 mm Hg. At perfusion pressures below 40 mm Hg, cerebral blood flow decreases abruptly, and it eventually ceases completely (67). Additionally, asphyxia or shifts in the brainstem or in the cerebral hemispheres that arise from the mass effects of a tumor can affect cerebrovascular autoregulation and induce a decrease in cerebral blood flow at perfusion pressures higher than 40 mm Hg.
Aside from these circulatory disturbances, an expanding tumor produces a variety of mechanical deformations of the brain, especially in adults or in older children whose sutures cannot widen and whose cranium cannot enlarge in response to a prolonged increase in intracranial pressure.
Clinically, the most significant of these mechanical deformations are the various herniations, notably herniation of the cerebellar tonsils through the foramen magnum, a feature of infratentorial tumors, and herniation of the hippocampal gyrus, particularly the uncus, down through the tentorial opening (Fig. 11.1). This herniation is observed with supratentorial masses, especially ipsilateral temporal lobe tumors.
Both types of herniations can produce secondary brainstem dysfunction, most probably by circulatory impairment and edema.
Clinical signs of occipital lobe infarction from increased intracranial pressure are less frequent. This condition results from kinking of the posterior cerebral arteries as they become wedged against the rigid tentorial edge (68). Because arterial pressure usually exceeds intracranial pressure, even in the presence of a mass lesion, local or systemic circulatory disturbances contribute to the development of arterial insufficiency.
Impaired cranial nerve function commonly accompanies increased intracranial pressure (69). The abducens nerve can become compressed between the anteroinferior cerebellar artery and the pons, producing weakness or paralysis of the lateral rectus muscle. This cause of sixth nerve paralysis was originally suggested by Cushing (70) and Collier (71) as a “false localizing sign.” The nerve also may be stretched as a result of transtentorial herniation, or it may be compressed directly against the clivus. Although its vulnerability to increased intracranial pressure has commonly been attributed to its length, anatomic studies have shown that the abducens nerve is approximately one-third the length of the trochlear nerve (71a). Involvement of the oculomotor nerve can accompany uncal herniations. The oculomotor nerve, particularly its pupillomotor fibers, can be compressed by the posterior cerebral artery. Pressure on the nerve can produce transitory circulatory disturbances or actual grooving of the nerve with distal petechial hemorrhages from the vasa nervorum.
Clinical Manifestations
Increased intracranial pressure produces headaches, vomiting, impaired vision, and changes in consciousness, and, when sutures have not fused, an enlarging head.
Headache
Headache, a constant feature of increased intracranial pressure in adults, is less common in children, being noted in 70% of the series of Bailey and coworkers (9), and in approximately 50% of children younger than age 13 years (72). However, headache is the most frequent presenting symptom in pediatric CNS tumors (73). Headaches from brain tumors result from traction on the dura and blood vessels at the base of the brain. Pain may be generalized or localized and worsen with exertion. In children, the site of the pain, most commonly bifrontal, rarely assists in localizing the site of the tumor. Posterior fossa tumors, however, can produce irritation of the posterior roots of the upper cervical cord, resulting in pain in the back of the head and neck. Stiffness of the neck and a persistent head tilt, which can be adopted to avoid diplopia, also indicate a mass in the posterior fossa. Most commonly, the headache is transitory, occurring in the morning or awakening the child during the night. In children younger than age 12 years, it can disappear temporarily with widening of the sutures, only to recur with further growth of the mass lesion. The majority of children who develop headache as a consequence of a brain tumor have it as their first symptom, though few have it as their only symptom. It has been reported that children with brain tumors have had headache for less than 4 months (95%) or 2 months (85%) (74,75). In a recent retrospective study of 200 children with brain tumors in the United Kingdom, 36% had headache for more than 4 months (73), and a dirunal pattern of headache was present in two-thirds. The longer the headache is present, the greater the likelihood of associated vomiting (77%), visual difficulties (57%), unsteadiness (45%), behavioral change (32%), and disturbed sleep (26%) (73). Thus, if headache has been present for 4 months or longer and the neurologic and ophthalmologic examinations are normal, it is unlikely the child has a brain tumor.
Vomiting
Vomiting is one of the most constant signs of increased intracranial pressure in children. It occurred in 84% of patients in the series of Bailey and associates (9), in 61% of children younger than 13 years of age in the 1975 series of Till (72), and in 51% in the series of Ferris (73). The reduced incidence of vomiting in the more recent series probably reflects an earlier referral. Vomiting can result from both increased intracranial pressure and from irritation of the brainstem by tumors of the posterior fossa. Contrary to general belief, vomiting in these patients is often not projectile, and it can be accompanied by nausea. Parents often note that vomiting relieves the headache. Like headaches, vomiting is most prominent in the morning and is unrelated to eating. Widening of the sutures can afford transitory relief.
Impaired Vision
Diplopia is usually the result of paralysis of one or both lateral recti muscles. Strabismus can be most striking at the end of a day and occasionally can fluctuate in parallel with the degree of increased intracranial pressure (Fig. 11.2). Visual problems were noted in 39% of children with brain tumors in Ferris’s series; the median duration of visual symptoms was 3 weeks. Cranial nerve abnormalities were the most common finding (49%) on neurological examination (73).
Papilledema
Bailey and coworkers noted papilledema in 80% of children with brain tumors (9), whereas in the later series of Till compiled in 1975, it was absent in 45% of children younger than 13 years of age (72). In the latest series of Ferris and coworkers, published in 2004 (73), it was
absent in 62% of children. This trend of decreasing prevalence of papilledema at the time of diagnosis may reflect more readily available neuroimaging modalities to evaluate headache. Papilledema represents an abnormal elevation of the nerve head. Obliteration of the disk margins and absent pulsations of the central veins are the earliest and most important ophthalmoscopic findings. When increased intracranial pressure has developed acutely, vascular congestion with hemorrhages and exudates also are conspicuous (Fig. 11.3).
absent in 62% of children. This trend of decreasing prevalence of papilledema at the time of diagnosis may reflect more readily available neuroimaging modalities to evaluate headache. Papilledema represents an abnormal elevation of the nerve head. Obliteration of the disk margins and absent pulsations of the central veins are the earliest and most important ophthalmoscopic findings. When increased intracranial pressure has developed acutely, vascular congestion with hemorrhages and exudates also are conspicuous (Fig. 11.3).
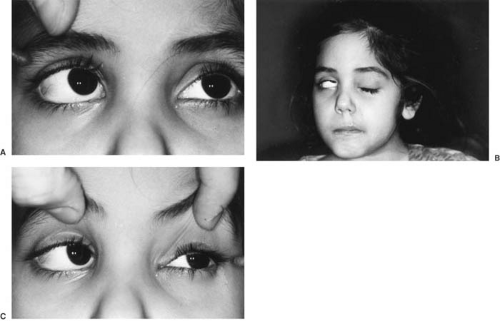 FIGURE 11.2. Right-gaze palsy (A) caused by involvement of paramedian pontine reticular formation and of the abducens nucleus itself is present in this child with brainstem glioma. As compared with intact left gaze (C), there is complete inability to move either eye to the right past the midline. Profound right peripheral facial paresis (B) is noted, with associated facial asymmetry and right orbicularis oculi weakness. |
Papilledema should be distinguished from pseudopapilledema. Pseudopapilledema is a congenital anomaly associated with small anomalous disks, less commonly with drusen (hyaline bodies) buried below the surface of the disks and resulting in excessive glial proliferation at the disk margins. The nerve fibers appear raised, creating a blurred disk with or without vascular tortuosity. This anomaly can be transmitted as a dominant trait, and frequently it accompanies hyperopia or congenital malformations of the skull, such as brachycephaly. The clinical diagnosis of pseudopapilledema is often difficult and rests on the absence of physiologic cups, and the normal central emergence of vessels, and most important, the absence of other signs of increased intracranial pressure (76,77). Although examination of the visual fields demonstrates a normal blind spot, in contrast to the increased blind spot
seen in true papilledema, this test is difficult to perform in young children.
seen in true papilledema, this test is difficult to perform in young children.
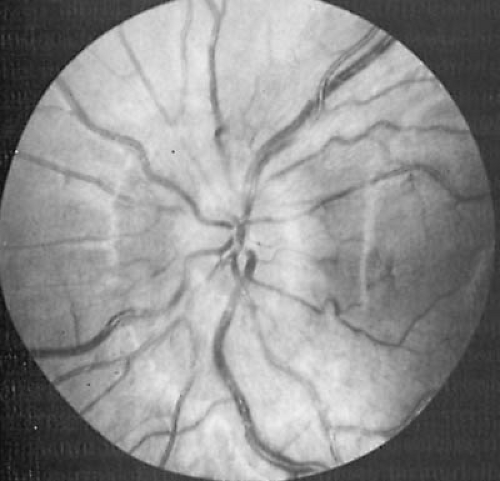 FIGURE 11.3. Papilledema in 10-year-old girl with pseudotumor cerebri. The disc margins are elevated, and the retinal veins are tortuous and distended. (Courtesy of Dr. Robert Hepler, Department of Ophthalmology, University of California, Los Angeles, UCLA School of Medicine.) |
Fluorescein fundus angiography has been advocated for distinguishing papilledema from pseudopapilledema. After the injection of fluorescein, the papilledematous optic disk discloses an increased capillary network, the presence of microaneurysms, and most significantly, persistence of fluorescence at the disk margins or over the edematous disk area. Generally, these changes are not observed in pseudopapilledema (78).
In papilledema, visual symptoms usually are insignificant or limited to transient attacks of blurred vision. The concentric enlargement of the blind spot, detected when charting of visual fields is possible, is always asymptomatic.
Increased intracranial pressure is an important factor in the production of papilledema, but other causes can contribute. Papilledema can occur with normal intracranial pressure, as in infectious polyneuritis. Conversely, it sometimes fails to appear despite a prolonged increase in intracranial pressure.
The most favored hypothesis to explain the mechanism for papilledema is that the subarachnoid spaces surrounding the optic nerves, which are continuous with those inside the skull, allow a free transmission of intracranial pressure. When pressure is elevated, optic nerve tissue pressure increases. If communication between the ventricular and the perioptic spaces is blocked, as occurs in some patients with hydrocephalus, then papilledema does not develop, even after a long-standing elevation of intracranial pressure. The increased pressure within the optic nerve in turn interferes with axoplasmic flow and causes stasis at the optic nerve head. This produces axonal swelling and the clinical appearance of papilledema. Compression of vessels over the surface of the disk results in their dilatation and in the formation of microaneurysms and hemorrhages. Obstruction of venous and lymphatic drainage is probably secondary to the increase in optic nerve pressure, rather than being its cause (77,79).
Although the presence of papilledema makes an intracranial mass highly probable, a number of other conditions must be excluded. In particular, choked optic disks also are encountered in pseudotumor cerebri, a condition characterized by increased intracranial pressure in the absence of a mass lesion.
Papilledema also can be found in the presence of subdural hematoma and in such systemic diseases as infectious polyneuritis, poliomyelitis, hypoparathyroidism, carbon dioxide retention, hypervitaminosis A, severe anemia, and lead poisoning.
Enlargement of the Head
Enlargement of the head is characteristic of a long-standing increase in intracranial pressure in infants and young children. In a series of brain tumors appearing during the first 2 years of life, an enlarging head was the most frequent presenting sign and was noted in 76% of infants (80). Enlargement of the head is not solely caused by brain tumors, but can occur in any child who has increased intracranial pressure and open sutures. Percussion of the head gives a high-pitched sound, termed cracked pot or Macewen’s sign.
TABLE 11.4 Location of Tumors Producing Seizures in Children | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Seizures
Seizures are rarely an early indication of mass lesion; in one series, they represented the initial symptom in approximately 15% of children with supratentorial lesions (81). Conversely, less than 1% of children with new onset seizures have a brain tumor as the etiology (82). Seizures become more frequent as the tumor grows and can be a late sign even in tumors of the posterior fossa. When seizures are caused by a brain tumor, patients rarely have other associated symptoms or signs of increased intracranial pressure. A complex partial seizure is the most common seizure type seen with intracranial masses, and the temporal lobe is the most common site for tumors producing seizures early in the course of illness (Table 11.4) (81).
Disturbances of Affect and Consciousness
Although mental disturbances or psychiatric disorders are fairly common with brain tumors in adults and indicate lesions of the frontal or temporal lobe, they are rarely observed in children. Children occasionally develop drowsiness, changes in personality, and irrational behavior, suggesting hypothalamic or thalamic involvement; changes in sleep patterns or in appetite are relatively common in frontal tumors. A progressive depression of consciousness resulting in drowsiness, stupor, and coma reflects increased intracranial pressure and compression of the reticular activating system. This compression can involve the reticular activating system in the upper brainstem when there is downward uncal or transtentorial herniation, or when, as a consequence of unilateral mass lesions, compression of the lower thalamus with horizontal displacement of the midbrain is seen (83).
Diagnosis
The diagnostic evaluation of the child suspected of harboring a brain tumor has undergone drastic changes since the 1970s. Classic texts, such as those by Bailey, Buchanan, and Bucy (9), published in 1939, bear witness to the early diagnostic difficulties presented by intracranial mass lesions. Formerly, initial evaluation involved the use of plain skull films and electroencephalography (EEG), followed by invasive procedures such as pneumoencephalography, ventriculography, and arteriography. This regimen has been superseded by various neuroimaging studies.
Diagnostic Neuroimaging
Advances in computer technology have helped to establish more effective methods of identifying and characterizing pediatric brain tumors (12,84). The primary objective for any diagnostic MRI or computed tomographic (CT) study is to distinguish between normality and abnormality, and to determine the relevance of the findings on neuroimaging to the clinical situation. Establishing normality is typically more difficult than recognizing obvious pathology. Once a lesion is observed, the role of imaging shifts toward more specialized tasks, including interpreting the nature of the lesion, establishing the spatial relationship between the mass and eloquent areas of the brain, and staging the extent of disease.
In the initial evaluation of a child with a brain tumor, careful staging is particularly essential to appropriate treatment decisions. This process is critical, as the initial surgical resection may be the only time some tumors can be cured. Therefore, it is imperative that the full extent of the lesion is appreciated. Additionally, cytoreductive surgical intervention may not be warranted if evidence exists of distant spread. Several factors contribute to assisting the radiologist with the differential diagnosis of a particular tumor: the age of the patient (Table 11.5), the location of the tumor (Table 11.6), and the inherent imaging features (Tables 11.7 and 11.8).
Unlike in adults, many masses in children are not actually tumors, and a variety of pathologic lesions can mimic the picture of a brain tumor on neuroimaging (Table 11.9). Some tumors are indolent, exhibiting virtually static biologic growth characteristics (85), whereas other mass lesions can display a mixture of both true tumor and cortical dysplastic features in the same region (86).
CT is used effectively for imaging the skull base, where the natural contrast of bone, air (paranasal sinuses), fat (nasopharyngeal/retrobulbar orbit), or CSF provides inherent contrast resolution. CT also is used whenever an imaging study is performed on an urgent basis. However, some tumors, especially those in the posterior fossa or temporal fossa, are best visualized with MRI and are occasionally missed by CT imaging (Fig. 11.4). This is particularly the case when the tumor is isodense and thus requires contrast infusion for its delineation (Fig. 11.5). In a brainstem glioma, for instance, sagittal scans demonstrate the rostrocaudal extent of the tumor (Fig. 11.6). Tumors involving the sella or the chiasmatic cistern are also more readily delineated by MRI than by CT because interference
from surrounding bone limits the accuracy of CT. In the experience of Sartor and coworkers, who compared the accuracy of CT scanning with MRI in delineating mass lesions in the sellar region, MRI was equivalent to CT scanning in 54% of cases, superior in 41%, and inferior in only 5% (87).
from surrounding bone limits the accuracy of CT. In the experience of Sartor and coworkers, who compared the accuracy of CT scanning with MRI in delineating mass lesions in the sellar region, MRI was equivalent to CT scanning in 54% of cases, superior in 41%, and inferior in only 5% (87).
TABLE 11.5 Pediatric Brain Tumors and Age at Presentation | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
TABLE 11.6 Pediatric Brain Tumors and Central Nervous System Location | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
TABLE 11.7 Magnetic Resonance Imaging Features of Benign Tumors | ||
|---|---|---|
|
Because CT imaging concepts are similar to standard radiologic acquisitions, and CT is less often required for neuroaxis tumor imaging, most of the following discussion focuses on MRI.
An understanding of the nature of a brain tumor and its location requires knowledge of T1, T2, and gadolinium-enhanced T1-weighted information (88). Unenhanced T1-weighted images can reveal areas of pathology that differ from normal brain by being either hyperintense or hypointense when compared with contiguous normal brain. Hyperintensity on T1-weighted images implies the presence of substances that naturally increase proton relaxation, such as methemoglobin (subacute residua of hemorrhage), melanin, dilute amounts of interstitial or intracellular calcification (postradiotherapy), and hyperconcentrated proteins (as might occur in a colloid-containing mass or hypercellular tumors). T1 hypointensity suggests changes related to either anisotropic effects (by structures containing either very large or rigidly bound molecules, as seen in intratumoral desmoplasia or matrix calcification) or, alternatively, tissue containing unbound water (as seen in peritumoral edema, necrosis, or cyst formation) (89,90). T1 hypointensity reflects both extremes of hydrogen states: too tightly bound or too loosely bound compared with protons in adjacent brain parenchyma.
TABLE 11.8 Magnetic Resonance Imaging Features of Malignant Tumors | ||
|---|---|---|
|
TABLE 11.9 Pediatric Brain Tumor Mimics | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Information from T2-weighted sequences differs from T1-weighted data. In physical terms, T2 rate reflects the time required for the signal to degrade or to disappear from the image. It is a measure of loss of coherence of the magnetic vectors that had been refocused by the second radiofrequency pulse in spin echo sequences or by gradient applications in gradient echo imaging. Tissue with faster T2 rates disperses its signal faster than tissue with slower T2 rates, and hence exhibits less signal intensity on the image. Tissue in which the water molecules are bound within large complex molecules or within rigid tissue has faster T2 rates, and therefore destroys its signal more quickly (T2 hypointensity). Abnormal T2 hypointensity implies a more restricted bulk water pool. The latter occurs in fibrosis, ossification, and dehydrated tissues. Tissues that
possess more unbound water molecules (such as CSF or vasogenic edema) have slower T2 rates and therefore exhibit a brighter signal. Abnormal T2 hyperintensity implies an increase in bulk tissue water.
possess more unbound water molecules (such as CSF or vasogenic edema) have slower T2 rates and therefore exhibit a brighter signal. Abnormal T2 hyperintensity implies an increase in bulk tissue water.
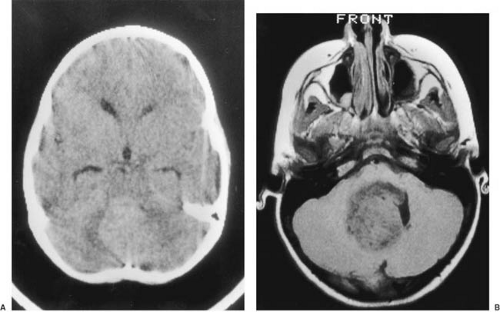 FIGURE 11.4. A: Computed tomographic scan of head shows ill-defined posterior fossa density that obliterates the fourth ventricle. B: Magnetic resonance imaging shows a well-defined midline posterior fossa tumor in the same patient. Magnetic resonance imaging is clearly superior to computed tomographic scanning in visualizing the tumor. |
For most intra-axial brain or spinal cord tumors, the T2-weighted sequences more accurately predict the extent of tumor infiltration. Although peritumoral tissue edema may not always contain tumor cells, the correlation is high. The zone of abnormality on standard spin echo T2-weighted images is a good measure of tumor infiltration.
Use of intravenously injected gadolinium (gadopentetate dimeglumine at 0.1 mmol/kg) or other paramagnetic compounds significantly improves the resolution of many brain tumors (86). T1-weighted, gadolinium-enhanced imaging is generally acquired in two orthogonal planes to ensure full appreciation of a tumor’s margins. The optimal planes of section, however, depend on the location of the tumor. Gadolinium enhancement arises whenever the capillary bed is expanded or vessels leak contrast through their walls. It assists in defining areas of edema and necrosis, cyst formation, spinal cord metastases, and extension of tumor to the leptomeninges or ependyma. High-grade tumors such as glioblastomas, ependymoblastomas, and PNET/MBs tend to enhance, whereas surrounding edema does not. Lack of enhancement increases the likelihood that the tumor is benign. MRI with gadolinium enhancement also is of considerable value in postoperative follow-up studies in distinguishing residual or recurrent tumor from edema, granulation tissue, and necrosis. As a rule, any structure that enhances with iodine on CT enhances on MRI with gadolinium; the reverse is not always the case. In a recent series of 225 patients by Laughton and colleagues (91), MRI linear enhancement following surgery almost always resolved or improved within 12 weeks. Increasing enhancing tissue at the surgical site correlated with local recurrence. The persistence of enhancing tissue beyond 12 weeks was usually due to residual tumor. The development of enhancing foci in the previously nonenhancing tumor was due to radionecrosis or histological change in the grade of the original tumor. Finally, nodular postsurgical enhancement was nonspecific and secondary to postsurgical changes, residual tumor, or occasionally, choroid plexus.
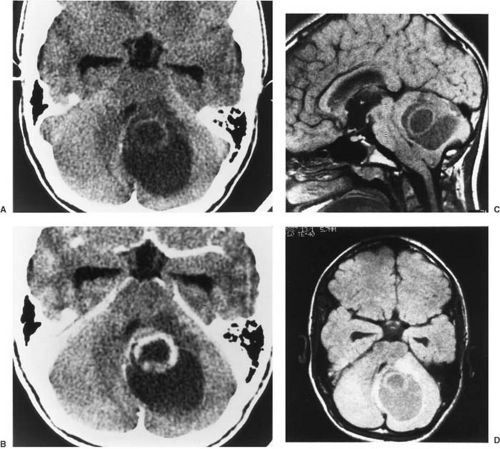 FIGURE 11.5. Computed tomographic scans revealing cerebellar cystic astrocytoma in a 13-year-old boy. Computed tomographic scans before (A) and after (B) injection of iodinated contrast material show a large lesion predominantly involving the left cerebellar hemisphere but extending to the vermis. The large, well-demarcated macrocyst contains proteinaceous fluid, which accounts for the high density of the cyst fluid compared with CSF in the suprasellar cistern, temporal horns, and third ventricle. A: A small fleck of calcium is noted medially within the wall of the macrocyst on the noncontrast computed tomography. An enhancing tumor nodule projects into the anterior portion of the macrocyst; some areas of the macrocyst wall are enhanced as well. T1-weighted magnetic resonance images in the sagittal (C) and axial (D) planes also are shown. These noncontrast magnetic resonance studies were done before gadolinium-diethylenetriaminepenta-acetic acid was approved for use in children. The magnetic resonance findings are concordant with the computed tomographic scan changes. If gadolinium-diethylenetriaminepenta-acetic acid could have been used, it probably would have enhanced abnormalities comparable with iodinated contrast on the computed tomographic scan. At surgery, a middle-grade astrocytoma was found. The macrocyst contained yellow fluid; small cysts and necrotic areas were found within the mural nodule. (Courtesy of Drs. Hervey D. Segall, Marvin D. Nelson, Jr., and Corey Raffel, Children’s Hospital, Los Angeles.) |
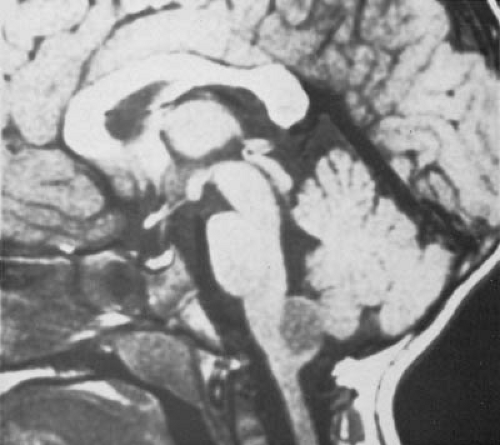 FIGURE 11.6. Sagittal T1-weighted magnetic resonance scan showing a posterior exophytic brainstem glioma in a 6-year-old girl. The scan shows a rounded, sharply marginated, low-intensity tumor projecting posteriorly from the medulla. This was a grade II astrocytoma. (Courtesy of Dr. Hervey D. Segall, Department of Radiology, University of Southern California School of Medicine.) |
Fluid-attenuated inversion recovery (FLAIR) sequences, which are now preferred by many radiologists to distinguish pathologic tissue edema in locations where CSF abuts brain, especially along the cortical surface and within the depths of sulci. Since fast scan techniques have been applied in conjunction with the FLAIR parameters, they have become the preferred imaging sequence in neuro-oncology. Other imaging techniques, including diffusion imaging, perfusion imaging, and functional imaging (such as MR spectroscopy) are increasingly used in neuro-oncology (92,93,94,95,96). Newer technologic developments are constantly unfolding to characterize tumor burden and the effects of tumor and therapies on brain structure and function (97,98). There is evidence that combining positron-emission tomography (PET) to image-guided surgical tumor resection provides contour to achieve total resection of ill-defined tumors and better resolution of residual disease following surgery (99,100,101). Methionine-PET is useful in distinguishing between benign and malignant CNS lesions (102). MRI diffusion, perfusion, and spectroscopy imaging tools are increasingly incorporated into clinical trials in pediatric neuro-oncology (103).
Other Studies
Ancillary studies are often used to further delineate the nature and the extent of the tumor. MR arteriography, or conventional arteriography using digital subtraction techniques, helps delineate the blood supply to the mass lesion (104). Additionally, these procedures can indicate the degree of malignancy and exclude the presence of a vascular malformation (105).
The dangers attending lumbar puncture in the presence of increased intracranial pressure preclude its use as part of the initial evaluation of a child with a suspected brain neoplasm. Because the presence and location of an intracranial abscess can readily be demonstrated by gadolinium-enhanced MRI, examination of the CSF is no longer used for this purpose. The appearance and composition of CSF in brain tumor patients were presented by Merritt in his classic study (106). Although CSF glucose level is almost always normal, it is reduced in some children with leukemic or lymphosarcomatous infiltration of the meninges and in meningeal carcinomatosis secondary to an ependymoma, a melanosarcoma, or a malignant tumor of the meninges (107).
Cytology can be performed on the CSF after centrifugation and can help determine whether the tumor has spread to the meninges or to the spinal subarachnoid space, as can occur in a PNET/MB (108).
The presence of malignant cells in the CSF indicates extensive seeding of the leptomeninges. In the experience of Glass and associates, such results were encountered only in patients with lymphomas and were the consequence of mistakenly identifying lymphocytes as lymphoma cells (108). False-negative results are fairly common in all types of tumors, with the exception of acute lymphatic leukemia (108). They are the result of the adherance of tumor cells to various structures of the CNS, such as the cauda equina, the cranial nerves, and the surface of the brain and the spinal cord (108a). In some instances, neuroimaging can show tumor spread to the CSF even when cytology is normal.
A less beneficial adjunct to the localization of an intracranial mass lesion is the EEG. Its greatest value lies in suggesting the presence of temporal lobe tumors in children with complex partial seizures. In such patients, the persistence of local slow-wave activity, overshadowing the spike and sharp-wave activity usually associated with seizures, points to a space-occupying lesion and calls for more definitive imaging studies.
Plain skull films can suggest an intracranial mass lesion if they show separation of sutures, erosion or enlargement of the sella turcica, or osteoporosis, especially thinning of the sphenoid ridge. Because the bones of the vault become more firmly joined, separation of sutures is rare in children older than 10 years of age and is practically nonexistent beyond age 20 years. In young children, sutures can spread after increased intracranial pressure lasting 10 days or even less, though suture separation takes longer to develop in older children (109). Radiographic evidence of calcification can be seen in 15% to 20% of intracranial tumors of childhood, mostly in the form of clusters of
multiple specks. Approximately 50% of oligodendrogliomas, a rare tumor in childhood, develop radiologic signs of calcification (110). The most common tumor to produce suprasellar calcification is the craniopharyngioma, and it does so in approximately 70% to 80% of cases, usually along the superior aspect of the mass. In tuberous sclerosis, multiple calcifications varying in size from specks to up to 1 cm are usually located along the ventricular walls, less often within the cerebrum and the posterior fossa.
multiple specks. Approximately 50% of oligodendrogliomas, a rare tumor in childhood, develop radiologic signs of calcification (110). The most common tumor to produce suprasellar calcification is the craniopharyngioma, and it does so in approximately 70% to 80% of cases, usually along the superior aspect of the mass. In tuberous sclerosis, multiple calcifications varying in size from specks to up to 1 cm are usually located along the ventricular walls, less often within the cerebrum and the posterior fossa.
The use of ultrasound in the detection of intracranial tumors is limited to neonates and infants with an open fontanelle (111).
Positron-emission tomography has considerable potential in delineating brain tumors. Primary brain tumors show good correlation between the uptake of labeled fluorodeoxyglucose (FDG) and the malignancy of the tumor as verified by its histology. Differences in uptake of FDG or labeled methionine also can assist in defining the site for a stereotactic biopsy (99,100,101). Additionally, the rate of FDG uptake after tumor irradiation generally allows one to differentiate between tumor recurrence, in which uptake is increased, and radiation necrosis, in which it is reduced (112). Corticosteroid therapy does not alter the PET scan. Thus, in the experience of Glantz and coworkers, 95% of patients with hypermetabolic abnormalities on the postoperative PET scan had recurrence of their tumor. Abscesses were, however, indistinguishable from recurrent tumors (113). The role of metabolic brain imaging as an adjunct to structural imaging is further discussed in the subsequent section dealing with radiotherapy.
Differential Diagnosis
Increased intracranial pressure is encountered not only in brain tumors, but also in various other mass lesions within the cranium. In infants, brain tumors cause increased intracranial pressure less commonly than do hydrocephalus, intracranial hemorrhage, and infections. Beyond 2 years of age, brain tumors become a progressively more frequent cause of increased intracranial pressure. Infections are encountered less often.
A brain abscess can act as a space-occupying lesion. It can arise from the direct spread of bacteria from an infected ear, mastoid, or paranasal sinus, or by hematogenous dissemination from a distant source. Brain abscesses are liable to occur in patients with congenital heart disease, particularly in those who have a significant right-to-left shunt. The presence of increased intracranial pressure in patients with congenital heart disease should arouse immediate suspicion of a brain abscess. Only approximately 30% of these children have fever, so its absence does not distinguish an abscess from a tumor. In most instances, MRI or CT scanning with contrast infusion is diagnostic of abscesses.
In pseudotumor cerebri, intracranial pressure is elevated in the absence of a mass lesion. No localizing signs are present, the spinal fluid is normal, and imaging studies show the ventricular system to be normal or smaller than normal, but not displaced or distorted.
In the past, one of the differential diagnoses was lead encephalopathy. This was particularly the case when generalized convulsions coincided with a clinical picture of a posterior fossa tumor (see Chapter 10).
The clinical differentiation of hydrocephalus of non-neoplastic origin from midline brain tumors usually requires MRI. Like hydrocephalus, mass lesions can produce striking cranial enlargement during the early months of life, and a gradually expanding periaqueductal tumor, such as a fibrillary astrocytoma of the quadrigeminal plate, can produce aqueductal stenosis and ultimately complete obstruction. MRI demonstrates not only the distortion of the ventricular system that can accompany a brain tumor, but also any small mass that might have produced aqueductal obstruction (114). Normally, CSF flow through the aqueduct produces a signal void on T2-weighted images (i.e., an area of blackness); absence of the void indicates cessation of flow and partial or complete obstruction.
Severe herpes simplex encephalitis can simulate a rapidly expanding temporal lobe lesion because of the massive edema that accompanies the infection and because of its predilection for the temporal lobe. Additionally, a small proportion of children with cerebral degenerative diseases, notably acute disseminated encephalomyelitis (ADEM) and subacute sclerosing panencephalitis, can develop increased intracranial pressure and papilledema. When these signs are accompanied by predominantly unilateral signs, as they frequently are, the clinical diagnosis of brain tumor is unavoidable and is refuted only by neuroimaging studies that demonstrate white matter edema (115).
Treatment
Surgical extirpation remains the mainstay of treatment for pediatric brain tumors. The advent of the operating microscope, laser beam, and ultrasonographic methods have made it possible to increase the number of children having safe and complete tumor resections.
Most patients harboring a brain tumor rapidly improve with the administration of dexamethasone at a 0.1 to 0.5 mg/kg starting dose. This reduces increased intracranial pressure and allows the surgeon a much less explosive environment in which to radically resect the tumor. Then, if necessary, the child should be immediately
transported to a brain tumor center. Other medications, such as phenytoin or fosphenytoin, can be used prophylactically in the perioperative period. The pediatric neurosurgeon is more than a technician in brain tumor surgery because the surgeon can provide vital information about extent of infiltration of the nervous system by direct observation of the brain tumor. Experience is the key to radical resection with low morbidity. The adaptation of the operating microscope to microneurosurgery, ultrasonic aspiration, and stereotaxis have made it possible to operate on brain tumors with minimal morbidity. The role of CT- and MRI-guided stereotactic biopsies in childhood brain tumors is appropriate and preferable to open biopsy in tumors involving the basal ganglia, thalamus, and midbrain. This simple, precise diagnostic test can provide an accurate diagnosis in approximately 95% of cases, with morbidity in the 2% to 5% range (116).
transported to a brain tumor center. Other medications, such as phenytoin or fosphenytoin, can be used prophylactically in the perioperative period. The pediatric neurosurgeon is more than a technician in brain tumor surgery because the surgeon can provide vital information about extent of infiltration of the nervous system by direct observation of the brain tumor. Experience is the key to radical resection with low morbidity. The adaptation of the operating microscope to microneurosurgery, ultrasonic aspiration, and stereotaxis have made it possible to operate on brain tumors with minimal morbidity. The role of CT- and MRI-guided stereotactic biopsies in childhood brain tumors is appropriate and preferable to open biopsy in tumors involving the basal ganglia, thalamus, and midbrain. This simple, precise diagnostic test can provide an accurate diagnosis in approximately 95% of cases, with morbidity in the 2% to 5% range (116).
The main goal of brain tumor surgery has remained constant: to maximize resection while preserving function. Patients with benign tumors such as pilocytic astrocytomas of the posterior fossa are often cured by complete resections. However, one of most common errors made during surgery is the discontinuation of a procedure when a tumor such as an astrocytoma, ependymoma, or PNET/MB is perceived to be emanating from the floor of the fourth ventricle (117). Commonly, these tumors involve the middle cerebellar peduncle adjacent to the fourth ventricle and should be totally resectable. For PNET/MB, it is widely accepted that the degree of tumor resection is important prognostically. Ependymoma can insinuate itself into an enlarged foramen of Luschka, but the surgical attempt will be discontinued inappropriately when the surgeon perceives the lesion to be invading the brainstem. Although the posterior fossa poses a considerable challenge to the neurosurgeon, new techniques such as intraoperative imaging with MRI are having a favorable impact on the safety of tumor resection (118). Whether such techniques will decrease the incidence of cerebellar mutism postoperatively remains to be seen.
Establishing the correct neuropathologic diagnosis of a pediatric brain tumor is critical in directing subsequent therapeutic efforts. Neuroimaging studies and perioperative frozen sections and touch preparations may be highly suggestive of a particular histologic type of tumor. However, a final diagnosis of surgically resected material often requires immunohistochemistry, flow cytometry, and electron microscopy. Therefore, it is best to wait 48 to 72 hours postoperatively for a final neuropathologic interpretation before discussing prognosis and any adjuvant treatment plans with the family.
Postsurgical Neuroimaging
The most important role of neuroimaging after surgery is to determine whether a mass has been completely resected and to assess the extent of any residual tumor. This information is most readily provided by serial imaging studies. Consequently, an immediate (24 to 48 hours) postoperative baseline study should be obtained. A variety of abnormalities can be observed. Postoperative edema can be present for days. Blood products can be seen for months. Tumor bed granulation tissue may be seen for years and can mimic residual tumor. MRI shows encephalomalacia, non-nodular contrast enhancement conforming to the encephalomalacia (granulation tissue), and hypodense susceptibility artifact in the same region as the enhancement on T2-weighted images. The reduced T2 signal is believed to be the result of microhemorrhages and hemosiderin deposition. Presence of nodular hyperintense abnormalities on T2-weighted sequences should raise the suspicion of residual neoplasia. Special attention to the depths of the middle fossa and to the cribriform plate is important because subarachnoid space extends to these regions and may harbor metastatic tumor.
Another surgically related abnormality is dural thickening and enhancement associated with the presence of an indwelling ventricular-peritoneal shunt catheter. The dural changes may occur near the entrance of the shunt or in a more diffuse fashion overlying the surface of the brain. Benign dural thickening is not nodular and generally ranges from 1 to 3 mm. The ventricular-peritoneal shunt typically reduces the volume of the ipsilateral lateral ventricle more so than the contralateral side. The value of surveillance brain imaging varies by tumor type. The purpose of serial imaging is to document the response to treatment and make a diagnosis of recurrence or complications as soon as possible (119).
Radiotherapy
Radiotherapy can be used either as an adjuvant to surgery or for definitive therapy when complete resection is not feasible. Standard techniques of pediatric CNS radiotherapy, such as limited-field radiotherapy and craniospinal radiotherapy, have offered curative therapy for low-grade gliomas, PNET/MB, germinoma, and other tumor types. Advanced radiotherapy techniques, including conformal therapy, fractionated stereotactic radiotherapy, and stereotactic radiosurgery, are improving survival and reducing neurotoxicity. Many factors must be considered in deciding precisely how to safely use radiotherapy.
Within cells, the most abundant molecule is water. In addition, lipids, proteins, carbohydrates, and nucleotides are present. When ionizing radiation interacts with a cell, reactive species are created that damage cellular DNA (120). Tumor cells are particularly vulnerable to such damage because of aberrant cell cycle control mechanisms and because radiotherapy technique (multiple beams of radiation) delivers more radiation to the tumor site than to normal brain.
For each histologically defined pediatric brain tumor, there are critical radiation treatment parameters, such
as total dose, time, and volume of tissue treated. Slow-growing focal tumors such as pilocytic and low-grade fibrillary astrocytomas may require limited-field radiation, whereas infiltrative tumors such as PNET/MB, germinoma, and atypical teratoid/rhabdoid tumor require craniospinal radiation.
as total dose, time, and volume of tissue treated. Slow-growing focal tumors such as pilocytic and low-grade fibrillary astrocytomas may require limited-field radiation, whereas infiltrative tumors such as PNET/MB, germinoma, and atypical teratoid/rhabdoid tumor require craniospinal radiation.
Another important factor in determining a treatment approach for pediatric brain tumors is whether radiation is used as a single modality or in its more common role as a component of multimodal therapy. Timing of treatment and the dose delivered are affected by adjuvant chemotherapy, surgery, or both. Furthermore, patient age and tumor location in relationship to dose-limiting critical neural structures significantly influence the anticipated sequelae of treatment.
Modern brain imaging machines have made the use of stereotactic radiotherapy techniques possible and have allowed for the delivery of large single doses of radiation to well-defined intracranial targets (121). In addition, radiation scatter is limited and thus, theoretically, should reduce late cognitive or endocrine effects from limited-field radiation of brain tumors. Advances in stereotactic treatment methods may soon supplant standard external-beam radiation, which has been used for decades (121).
In models of external-beam radiotherapy, treatment planning begins with construction of a face mask for reproducible position. Using this system along with standard relocation technologies such as laser beams, a patient can be repositioned daily with an accuracy of 3 to 9 mm (122). Stereotactic radiosurgery requires placement of a stereotactic head ring, which is attached to the patient’s skull. Imaging in reference to the ring can then identify mathematical coordinates for each pixel in the CT or MRI images that directly corresponds to points in the patient positioned in the ring. Treatment planning identifies a coordinate at the center of a treatment plan, and data can be transferred to a treatment delivery machine with high accuracy (123). The technique uses the equivalent of hundreds of radiation beams delivered with circular fields via multiple arcs of treatment. This allows the radiation dose to be delivered accurately and to fall off optimally outside the treatment volume. Although a large single-dose treatment has radiobiologic disadvantages for more malignant tumors, the advantage of accurate avoidance of normal tissue with steep dose gradients has made this a proven therapy for several entities, including arteriovenous malformations ependymomas, acoustic schwannomas, metastases, and meningiomas (124,125).
Use of stereotactic radiosurgery in benign and malignant tumors in children has been less well defined than in adults, although experience in pediatrics is increasing. Stereotactic radiosurgery has been a valuable adjunctive strategy in the management of recurrent or unresectable pediatric astrocytomas (126). Further details of this procedure are provided by Bova and coworkers (127), Flickinger and coworkers (128), and Murthy and colleagues for posterior fossa tumors (129). Proton radiation therapy enables the radiation oncologist to conform the radiation dose around even highly irregular tumor volumes with homogeneous dose distribution throughout. Hug and colleagues have reported use of conformal proton radiation therapy for pediatric low-grade astrocytomas (130). Proton radiation therapy may also allow reduction of the cochlear dose of radiation associated with sensorineural hearing loss that is a significant late effect from treatment of brain tumors (131).
Immediate and Long-Term Effects of Radiation Therapy
As improved surgical and radiation techniques have lengthened survival of children with brain tumors, concern has grown about the quality of life in survivors and about the nature and extent of radiation-induced deficits.
Pathologic and experimental studies indicate that the major pathophysiology of radiation-induced brain damage results from damage to the vascular endothelium, tissue that is more radiosensitive than neurons. This results in obstruction of small and medium-sized blood vessels with ensuing thrombosis and infarction of deep white matter, but relative sparing of cortex and subcortical white matter. Large vessel injury is far less common (132). Additional or alternative mechanisms of brain injury have been proposed. These involve direct injury to glia and axons, and, less likely, an autoimmune vasculitis (133). Vasogenic edema is probably responsible for acute reactions that develop within a few weeks of radiation.
Neurologic symptoms can develop during radiation, or shortly thereafter, or have their onset after an interval of many months or years.
Acutely, craniospinal radiation may produce nausea and vomiting within 2 to 6 hours of treatment because the radiation beam exits through the stomach and bowel. In contrast, some children exhibit anticipatory nausea and vomiting that occurs before or within 2 hours of treatment. Symptoms are typically well controlled with antiemetics such as prochlorperazine, but occasionally require newer, more potent agents such as ondansetron.
Several weeks after the start of radiotherapy, patients lose hair and complain of skin soreness, erythema, and sore throat. These side effects begin to abate 1 to 2 weeks after therapy. Use of topical cream should be avoided unless the radiation oncologist is consulted because many are oil-based and contain metal, which increases the surface radiation dose during therapy. After therapy is complete, aloe-based creams are recommended, and occasionally corticosteroid-based creams or newer matrix gels are used. Craniospinal radiation can cause thrombocytopenia, leukopenia, and anemia; thrombocytopenia delays completion of radiation treatment. Thus, complete blood counts should be obtained weekly during radiotherapy. If a delay is necessary, any limited-field external beam boost
or fractionated stereotactic approaches may be used in the interim. These regionally limited treatments allow bone marrow recovery without placing the patient at risk for future tumor progression.
or fractionated stereotactic approaches may be used in the interim. These regionally limited treatments allow bone marrow recovery without placing the patient at risk for future tumor progression.
Acute radiation encephalopathy occurs in the course of treatment whenever large daily fraction doses are administered. The underlying pathology is that of cerebral edema caused by vascular damage and an ensuing increase in vessel wall permeability, which in turn produces perivascular edema, and in the most severe cases, microhemorrhage and thrombi. Cortical neurons disappear, and fibrillary gliosis extends from the cortical surface into the deeper layers of gray matter. Radiation injury to brain parenchyma results in intravascular and perivascular microcalcifications. These are most common at the junction of cortical gray and white matter and the basal ganglia (134).
The clinical picture varies. In general, it consists of lethargy, nausea and vomiting, seizures, and an exacerbation of pre-existing neurologic deficits. MRI shows focal, multifocal, or diffusely increased signal in white matter on T2-weighted images (135). Lesions tend to start as small foci that progress with time. They are irreversible and do not correlate well with neurologic or developmental deficits (136). Acute radiation encephalopathy can be treated with corticosteroids, but it is preferable to prevent complications by administering low daily dosages, particularly in the rare instance in which radiotherapy is considered essential in infants and young children.
During therapy, some children are fatigued and do not make optimal effort in cognitive testing. Thus, it is best to complete radiation treatment before assessing cognition. Fatigue usually diminishes weeks to months after therapy, although many patients experience an early delayed response termed the somnolence syndrome in the 6 weeks to 3 months after radiotherapy.
The late effects of radiotherapy include impaired cognition, defective endocrine function, and the development of a second neoplasia.
A reduction of cognitive function is encountered in children irradiated for supratentorial and infratentorial tumors (120). Retrospective studies indicate that between 10% and 80% of surviving children who have undergone either local or whole-brain radiation therapy demonstrate a significant loss in full-scale IQ (134). Craniospinal radiation can be expected to lower IQ by 10 to 20 points (137). In a study by Spiegler and colleagues, results showed a 2- to 4-point decline per year in intelligence scores, with more rapid declines over the first few years (138). In addition, almost all children who have retained a normal IQ can be found to have some degree of attention and memory deficit (137). Visual perceptual skills, learning abilities, memory, and adaptive behavior also are adversely affected (138,139,140). These cognitive deficits are most evident in children younger than 7 years of age and appear to progress over the course of several years after completion of radiotherapy (141,142). Infants younger than 3 years of age have the worst prognosis in terms of intellectual development (143,144). Adjuvant chemotherapy, particularly intraventricular or intrathecal methotrexate, is believed to increase the risk for the development of dementia, as does a tumor localized to the cerebral hemispheres or the hypothalamus (137,145). Thus, the commonly used regimen of cranial irradiation and methotrexate can induce a necrotizing encephalopathy several months to years after completion of treatment. The incidence of necrotizing encephalopathy is dose related with respect to methotrexate and irradiation. However, children under age 10 years with brain tumors treated without radiation have a stable IQ (146). It is too early to know if hyperfractionated and reduced boost radiotherapy will decrease cognitive effects in children with PNET/MB without increasing the risk of tumor recurrence. Children with ependymoma who receive local radiotherapy are at much lower risk for worsening cognition, but postoperative neurological deficits are strongly correlated with poor intellectual outcome (147).
Neuroimaging studies in children with reduced cognition demonstrate white matter damage. On MRI, increased signal is seen in the periventricular white matter on FLAIR or T2-weighted images. The involved area has a scalloped appearance; often it extends to the gray-white matter junction (148). Zimmerman and colleagues have graded the white matter changes observed on MRI (149). There appears to be a direct relationship between the volume of brain irradiated and the development of white matter changes (150); the correlation between the severity of white matter involvement and the neurologic and cognitive deficits is not as good. This is particularly true for young children whose abnormalities on MRI tend to be less pronounced than those of older children (151). A longitudinal MRI study showed a decline in corpus callosum volumes in posterior subregions that receive the highest total dose of irradiation in treatment of PNET/MB (152).
Because of these sequelae, hyperfractionated approaches to treatment have been designed for treatment of the youngest children who must receive radiotherapy for tumor control. Careful attention to obtaining baseline studies before beginning radiotherapy is essential, as is recognition that the tumor itself plus surgery, chemotherapy, and psychosocial factors all contribute to function and coping.
A second important late effect of irradiation of the craniospinal axis is a disturbance in various growth and endocrine functions. This is seen in some 70% to 90% of patients (144,153). Although acutely not problematic, pituitary function can decline with time in 50% to 70% of patients, depending on the radiation dose and fractionation used. As a rule, the hypothalamus is more sensitive to radiation than the pituitary, and growth hormone secretion is affected first (134). An irreversible growth
hormone deficiency becomes apparent 1 or more years after completion of irradiation and produces a gradual diminution of growth rate (154,155). In the series of Constine and coworkers, only 36% of children who received cranial irradiation, and 25% of children who received craniospinal irradiation, attained a height greater than the third percentile (153). Irradiation of the spine significantly affects the overall crown-rump height of a young patient. Furthermore, the musculature and size of the vertebral bodies and sternum may be affected by treatment. These growth anomalies yield a characteristic body habitus (120). In addition, cranial treatment in young children can affect skull growth. These growth anomalies are important to recognize because musculoskeletal function and appearance are important determinants of self-esteem, coping, and social function in children and adolescents. Gonadotropin deficiency with delayed or precocious puberty also is encountered. Seventy percent of irradiated postpubertal girls had oligomenorrhea, and 30% of postpubertal boys had low testosterone levels. Symptoms of hypothyroidism were seen in 28%, and 62% had low total or free serum thyroxine levels (153). Interestingly, a recent report from Massimino and colleagues showed that thyroid stimulating hormone (TSH) suppression through I-thyroxin medication had protective activity on thyroid function at three years following radiotherapy for PNET/MB (156). Diabetes insipidus owing to irradiation has not been reported (154). In a recently reported series of survivors of pediatric brain tumors (a mix of patients having had surgery, radiotherapy, and chemotherapy), 85.3% had hypothyroidism, 73.5% had proven/probable growth hormone deficiency, 44.1% had adrenal insufficiency, 38.2% had hypogonadism, 34% had diabetes insipidus, and 8.8% had precocious puberty; 32.4% of patients with neuroendocrine sequelae had panhypopituitarism (157). However, in a study by Spoudeas and colleagues of children with posterior fossa tumors, there was a low incidence of anterior pituitary hormone deficiencies (158). Regrettably, radiation to the hypothalamic-pituitary axis and/or whole brain radiation seems to have little effect on the high prevalence of body mass indexes above the 95th percentile in children with brain tumors (31.5%) (159).
hormone deficiency becomes apparent 1 or more years after completion of irradiation and produces a gradual diminution of growth rate (154,155). In the series of Constine and coworkers, only 36% of children who received cranial irradiation, and 25% of children who received craniospinal irradiation, attained a height greater than the third percentile (153). Irradiation of the spine significantly affects the overall crown-rump height of a young patient. Furthermore, the musculature and size of the vertebral bodies and sternum may be affected by treatment. These growth anomalies yield a characteristic body habitus (120). In addition, cranial treatment in young children can affect skull growth. These growth anomalies are important to recognize because musculoskeletal function and appearance are important determinants of self-esteem, coping, and social function in children and adolescents. Gonadotropin deficiency with delayed or precocious puberty also is encountered. Seventy percent of irradiated postpubertal girls had oligomenorrhea, and 30% of postpubertal boys had low testosterone levels. Symptoms of hypothyroidism were seen in 28%, and 62% had low total or free serum thyroxine levels (153). Interestingly, a recent report from Massimino and colleagues showed that thyroid stimulating hormone (TSH) suppression through I-thyroxin medication had protective activity on thyroid function at three years following radiotherapy for PNET/MB (156). Diabetes insipidus owing to irradiation has not been reported (154). In a recently reported series of survivors of pediatric brain tumors (a mix of patients having had surgery, radiotherapy, and chemotherapy), 85.3% had hypothyroidism, 73.5% had proven/probable growth hormone deficiency, 44.1% had adrenal insufficiency, 38.2% had hypogonadism, 34% had diabetes insipidus, and 8.8% had precocious puberty; 32.4% of patients with neuroendocrine sequelae had panhypopituitarism (157). However, in a study by Spoudeas and colleagues of children with posterior fossa tumors, there was a low incidence of anterior pituitary hormone deficiencies (158). Regrettably, radiation to the hypothalamic-pituitary axis and/or whole brain radiation seems to have little effect on the high prevalence of body mass indexes above the 95th percentile in children with brain tumors (31.5%) (159).
Replacement of growth, thyroid, adrenal, and sex hormones should be overseen by an endocrinologist (160,161). No conclusive data show an increased risk of tumor recurrence with growth hormone replacement. As an extra precaution, however, it is common practice for endocrinologists to ensure that there has been no residual tumor or evidence of tumor progression for at least 2 years before starting growth hormone therapy. In addition, many recommend more frequent brain imaging in the first year of growth hormone therapy.
An uncommon but devastating late effect is a second unrelated malignancy within or outside the CNS (162,163,164). It is estimated that one in six children who have had a brain tumor develop this complication years after radiation (165,166). A different type of cancer found within a radiation field at a significant interval after completion of treatment is considered a radiation-induced malignancy (164). In the series of Meadows and colleagues (153), 68% of tumors developed in radiation-exposed tissue. Tumors result not only from prior radiation, but also reflect a genetic predisposition to malignancies. These second cancers can be highly malignant and respond poorly to available therapies (162,167).
Children treated with high-dose radiation also are at a ninefold increase in risk for developing second, unrelated CNS tumors. These appear at intervals of years to decades after irradiation and are often highly malignant (134). In a study at St. Jude Children’s Research Hospital (168), the 15-year cumulative incidence of second neoplasm and second malignant neoplasm was 5.3% and 4%, respectively (168). Young patients and patients with choroid plexus tumors appear to have an increased risk of second neoplasms that is associated with genetic factors (169).
Late radiation encephalopathy is a much less common complication. It develops several months to years after completion of treatment and is caused by brain necrosis or atrophy. The clinical picture is highlighted by headaches, seizures, and focal neurologic signs, notably hemiparesis. The course is slowly progressive. Imaging studies demonstrate low-density lesions within the brain. Symptomatic intraparenchymal hemorrhages outside the primary brain tumor site also have been recorded as a late complication (170). The exact cause of late radiation encephalopathy is unknown, but it is believed to result from a large total radiation dosage or large daily fractions (171).
Rarely, one observes a transitory episode of demyelination that appears some 2 to 3 months after completion of radiation and slowly resolves over the course of 6 to 8 weeks.
A late-onset myelopathy is another rare complication. Symptoms can be acute or chronically progressive. They range in severity from mild sensory signs, or lower motor neuron disease, to an acute paraplegia or quadriplegia (172). Cerebrovascular disease, manifested by strokes or transient ischemic attacks, can develop years to decades after completion of radiotherapy, particularly when radiotherapy is combined with chemotherapy. Damage to medium or large vessels—-in particular, the carotid artery and its major branches—-often can be documented (173). Recurrent attacks of migraine-like headaches lasting 2 to 24 hours and associated with focal symptoms such as aphasia or hemiparesis can represent a late complication of irradiation and chemotherapy (174). Children respond to propanolol or other antimigraine medications. Cerebral arteriography can trigger such an attack in these children.
In view of the high incidence of serious complications, there has been a trend toward delaying radiotherapy in
patients younger than 3 years of age; prolonged postoperative chemotherapy is the preferred treatment. Such a regimen appears to be well tolerated, and its results compare favorably with those obtained in children treated with postoperative radiation and adjuvant chemotherapy. It appears, therefore, that postponement of radiation therapy is a feasible alternative for small children and infants (175).
patients younger than 3 years of age; prolonged postoperative chemotherapy is the preferred treatment. Such a regimen appears to be well tolerated, and its results compare favorably with those obtained in children treated with postoperative radiation and adjuvant chemotherapy. It appears, therefore, that postponement of radiation therapy is a feasible alternative for small children and infants (175).
 FIGURE 11.7. T1-weighted coronal magnetic resonance imaging with gadolinium demonstrates deep white matter enhancing lesion in right hemisphere (A). 99mTc HMPAO uptake (B) was decreased and 201Tl uptake was absent (D). Histologic preparation (C) reveals coagulative necrosis, consistent with radiation effect, and no evidence of tumor (hematoxylin and eosin, original magnification x66). The patient was originally thought to have had a low-grade glioma and thus received radiotherapy. The patient is free of tumor more than 8 years from diagnosis. |
Neuroimaging After Radiotherapy
Neuroimaging can disclose a variety of changes consequent to radiotherapy. Most commonly, one sees mucositis within the paranasal sinuses and mastoid air cells and replacement of fat in marrow space. T2 hyperintensity may persist for months after stereotactic radiotherapy or radiosurgery. Standard-dose craniospinal radiotherapy produces few acute or subacute changes. Patients receiving whole-brain or craniospinal radiation may, after a period of several months, develop mild diffuse brain or spinal cord atrophy and chronic ischemic demyelination.
Radiation dosages that exceed neural tolerance produce radiation necrosis or progressive necrotizing leukoencephalopathy. The latter is usually seen when radiation therapy is used in conjunction with chemotherapy. The appearance of radiation necrosis on MRI and CT studies mimics residual or recurrent tumor (Fig. 11.7). Metabolic imaging tools such as 2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography, or thallium 201 single-photon emission CT (SPECT) can characterize metabolic activity within lesions (176,177,178,179). Neoplastic lesions have increased uptake of FDG and thallium, whereas radiation necrosis has decreased FDG metabolism as compared with normal surrounding brain; no thallium uptake occurs on SPECT imaging (179). Evidence exists that thallium is a
more sensitive marker of viable tumor, whereas FDG provides information about metabolic activity in brain surrounding tumor (179). Despite these advances in metabolic characterization of lesions that arise after radiotherapy, biopsy is still the diagnostic gold standard.
more sensitive marker of viable tumor, whereas FDG provides information about metabolic activity in brain surrounding tumor (179). Despite these advances in metabolic characterization of lesions that arise after radiotherapy, biopsy is still the diagnostic gold standard.
Chemotherapy
One of the concerns about using chemotherapy in treating brain tumors has been the fact that chemotherapy drugs cannot penetrate the blood–brain barrier with ease. However, blood vessels within tumors, particularly malignant lesions, are much more permeable than normal vessels, thus allowing entry of drugs. The heterogeneity of tumors and their vasculature probably accounts for incomplete entry of drugs in different regions of a mass lesion; invading tumor cells at a distance from the primary mass are less likely to encounter cytotoxic chemotherapy because surrounding vessels retain their blood–brain barrier. Despite these limitations, clinical trials have shown safety and efficacy of chemotherapy in pediatric brain tumors, such as PNET/MB, germinoma, and pilocytic astrocytoma, and in infants with various malignant lesions. To overcome the blood–brain barrier, strategies have been designed to concentrate chemotherapy agents within brain tumors. One of the proposed strategies is to implant a pump that administers chemotherapy. The pump is a self-contained delivery system connected to the brain or spinal cord. Although pumps have the capacity to deliver drugs more uniformly than pills or injections and to bypass the blood–brain barrier, their overall effectiveness for managing brain tumors remains inconclusive (180).
Another approach is the use of polymer matrixes (plastic) to improve local therapy of malignant brain tumors. The new plastics enable the drugs to diffuse into the tumor bed in a continuous, uniform fashion. The plastic casings also protect unstable drugs from the body’s own enzymes that may prematurely break them down. The pliability of plastics enables them to be molded into a variety of shapes and sizes that can be surgically implanted or injected into precise locations in the body (180). Langer and colleagues demonstrated that polymer systems can be used to treat malignant adult brain tumors (180). Carmustine (BCNU)–containing polymer disks have been implanted to deliver tumor-killing doses of BCNU even after 4 weeks. Preliminary evidence suggests that implanting a BCNU polymer disk into the area where a tumor has been resected reduces the recurrence rate of such tumors (180,181). No studies have as yet tested pumps or polymer systems in malignant pediatric brain tumors.
Another option has been the use of chemotherapy in conjunction with drugs such as mannitol to disrupt the blood–brain barrier and thus permit better access of systemic chemotherapy to tumor cells. Preliminary experiments have shown that this approach results in a high incidence of seizures and other complications without demonstrating evidence of better tumor control. The approach has not been tested for safety or efficacy in children with malignant brain tumors.
It has become clear that chemotherapy increases disease-free survival in some children (78). As a consequence, by using chemotherapy, radiation can be delayed and neurotoxicity ameliorated in many infants (182). Chemotherapy can reduce the size of low-grade glioma, optic glioma, and oligodendroglioma (183). High-grade glioma and ependymoma, however, are relatively chemoresistant (184). It is important to recognize that the chemosensitivity of brain tumors varies greatly among tumor types and among patients with a particular histologic type. High-dose chemotherapy with autologous stem cell rescue has increasingly been used in clinical trials to improve the dismal prognosis of young patients with malignant supra and infratentorial tumors (185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200). A discussion of the safety, toxicity, and efficacy of high-dose chemotherapy in brain tumors is far beyond the scope of this text. This area has been reviewed by Wolff and Finlay (200a).
Chemotherapeutic agents are toxic not only to malignant cells, but also to normal ones (201). Because they are carried out by the blood circulation, they can affect any tissue in the body. Most drugs have preferential activity for rapidly proliferating cells such as hair follicles (alopecia), bone marrow (anemia, thrombocytopenia, granulocytopenia), intestinal mucosa, and gonads. In many cases, the effects are temporary when used in conventional doses. The spectrum of systemic side effects from chemotherapy is dependent on agent and dosage (202).
Almost all agents cause transient alopecia, which begins within a month of starting treatment and resolves when treatment is discontinued. Chemotherapy can affect all three hematologic cell lines. Anemia and thrombocytopenia may require transfusions. Weekly blood counts are indicated in most cases. Nausea and vomiting are common within 4 to 5 hours of administration and may persist for a few days. These symptoms are best controlled with the serotonin antagonist ondansetron. Stomatitis, glossitis, and proctitis occur with the beginning of neutropenia and disappear once the blood count recovers.
Vincristine frequently causes transient constipation and jaw pain (203). Virtually all patients develop hyporeflexia or areflexia. Less common side effects include foot drop, wrist drop, cranial nerve palsies, and vocal cord paralysis. Seizures with hyponatremia and inappropriate secretion of antidiuretic hormone can occur. Cisplatin can produce irreversible hearing loss in the high-frequency range. It also can cause severe hearing loss in the normal speech frequency range, especially if combined with radiotherapy. The dose-limiting toxicity of cisplatin is nephrotoxicity, and most patients ultimately have a permanent reduction of the glomerular filtration rate. More complete
and detailed accounts of the systemic and neurotoxic complications of chemotherapy alone or in combination with radiotherapy are discussed in several excellent texts and reviews (204,205).
and detailed accounts of the systemic and neurotoxic complications of chemotherapy alone or in combination with radiotherapy are discussed in several excellent texts and reviews (204,205).
Neuroimaging After Chemotherapy
Chemotherapy can produce a variety of changes in the brain, ranging from mild atrophy to a destructive encephalopathy. The most severe complication of chemotherapy, usually seen in tandem with radiotherapy, is progressive necrotizing leukoencephalopathy (PNL) (206). PNL is characterized on MRI and CT as multicentric areas of contrast enhancement scattered randomly throughout cerebral white matter. The sites of the lesions do not necessarily correspond anatomically with the site of the primary tumor or the epicenter of radiotherapy. The findings may be more intense along the tract of an Ommaya reservoir catheter, presumably because of a higher concentration of chemotherapy from intraventricular injections. In time, the lesions calcify, and their widespread presence is associated with diminished IQ, neurobehavioral disturbances, and neuroendocrine sequelae (Fig. 11.8).
Brain changes have been associated with the use of specific chemotherapeutic agents (e.g., cyclosporine) that have been given intravenously rather than intrathecally. These agents can infrequently produce a zone of infarction in deep capillary beds. For the most part, these have occurred in the parieto-occipital regions and produced vision loss. The mechanism is presumed to be related to a regional vasculopathy, platelet activation, and capillary zone infarction.
 FIGURE 11.8. Computed tomographic scan in axial plane shows widespread calcifications in both cerebral hemispheres resulting from radiotherapy and chemotherapy treatment. |
Cutting-Edge Therapies
Advanced therapies for brain tumors are being designed to block tumor cell-surface receptors, inhibit signal transduction, inhibit tumor infiltration and metastasis, or inhibit the angiogenesis that is so essential to tumor survival. For example, therapeutic agents that inhibit the activation of tyrosine kinase receptors that mediate tumor growth (EGFR, HER2) and survival (TrkA, IGF1) are being developed. The primary challenges to developing effective targeted molecular therapies include the heterogeneity of tumors and their variability of receptor expression, limited specificity of agents for targeted receptors, and multiple pathways to signal transduction.
One of the more compelling approaches is that of targeting microvessels in evolving tumors. As tumors progress, they have redundant growth factor capacity; such growth factors such as bFGF and aFGF are more dominant in pediatric tumors during progression (207). Thus, agents that would modulate relevant growth factors and genes that contribute to angiogenesis in pediatric brain tumors could be highly effective. Over 30 antiangiogenesis agents are in clinical trials in the United States (approximately 70 worldwide) for treatment of a variety of malignancies. So far, there are no studies reporting results from use of antiangionesis agents in pediatric brain tumors. Gene therapy has been one of the most intellectually satisfying approaches. This modality offers hope for ultimately treating the root cause of the problem by replacing defective genes, amplifying the immune response to neoplasia, and sensitizing tumor cells to systemic therapies (suicide gene therapy).
Gene therapy involves the transfer of one or more genes and the sequences controlling their expression into target cell populations. Theoretically, gene therapy replaces defective genes, alters the immune response, and inserts drug-sensitizing genes. Currently, the most efficient delivery systems to transfer genes intracellularly are viral vectors such as adenovirus, adeno-associated virus, and retrovirus.
Insertion of the thymidine kinase gene from herpesvirus into tumor cells can sensitize them to intravenous ganciclovir (208,209). A replication-incompetent retrovirus (murine leukemia virus) contained within vector-producer cells (fibroblast vector-producer cells) is used to deliver thymidine kinase gene from herpesvirus. This gene therapy strategy is the most mature innovative approach and has been tested in vitro, in animals, and in a series of adult and pediatric clinical trials (209). Pivotal to the thymidine kinase gene from herpesvirus strategy is the bystander effect, which results in a larger number of tumor cells being killed than those that have been genetically altered.
Immunocompetence and the presence of gap junctions between tumor cells are required experimentally to obtain the bystander effect. The various hurdles inherent to the strategy of gene therapy are reviewed by Maria and colleagues (208,209,210). Limited clinical studies conducted in children with recurrent brain tumors have not shown an improved outcome with the various newer treatment modalities.
Immunocompetence and the presence of gap junctions between tumor cells are required experimentally to obtain the bystander effect. The various hurdles inherent to the strategy of gene therapy are reviewed by Maria and colleagues (208,209,210). Limited clinical studies conducted in children with recurrent brain tumors have not shown an improved outcome with the various newer treatment modalities.
For the time being, however, surgery, radiotherapy, and chemotherapy continue to be the mainstays of pediatric brain tumor management, and despite the many promising approaches, the problem with treating deep or inaccessible tumors still remains.
SPECIFIC TUMOR TYPES
There have been many attempts to classify CNS tumors based not only on traditional morphology, but also on their biologic, immunopathologic, and genetic features. The most recently updated classification was published by the World Health Organization in 1993 and was based, whenever possible, on the histiogenesis of tumors and their biologic behavior (211,212,213,214). Because the same type of tumor can produce totally different clinical pictures depending on its site, this chapter, for the greater part, adheres to a location-based classification. A standardized method for grading adult neuroglial tumors is based on the presence or absence of four histologic features: endothelial proliferation, necrosis, mitosis, and nuclear atypia. This scheme has been determined to be inappropriate for assesssing childhood supra- and infratentorial neuroglial tumors and adds little in terms of prognostic information (214a,214b).
Pediatric brain tumors include a spectrum of both glial and nonglial tumors that differ significantly in location and biologic behavior from those found in adults. Brain tumors in infants and children most often arise from central neuroepithelial tissue, whereas a significant number of adult tumors arise from CNS coverings (e.g., meningioma) or adjacent tissue (e.g., pituitary adenoma) or are metastases.
Tumors of the Posterior Fossa
Cerebellar Tumors
The cerebellum can be the site of various neoplastic processes, three of which are particularly common in children. These are the cerebellar pilocytic astrocytoma, a generally benign tumor arising from the cerebellar hemispheres or vermis, the PNET/MB, a malignant invasive tumor arising from the fetal cerebellar granular layer of the anterior and posterior medullary vela, and the ependymoma, which arises from the ependymal layer of the fourth ventricle (215). The incidence of the various neoplasms in the infratentorial compartment is shown in Table 11.10 (216).
TABLE 11.10 Frequencies of Common Pediatric Brain Tumors in the Infratentorial Compartment | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Pathology
Astrocytomas.
These tumors are derived from the astrocytic neuroglial cells and can arise from either the vermis or the lateral lobes of the cerebellum. They are almost always well circumscribed and tend to be cystic, with the neoplasm confined to a small intramural nodule (see Fig. 11.9). Occasionally, astrocytomas form a solid tumor mass involving the cerebellum, vermis, and their brainstem connections.
The microscopic appearance of cerebellar astrocytomas varies considerably even within the same tumor. On the basis of histologic characteristics, Gilles and his group distinguished two tumor types whose resectability and long-term survival can be predicted (216,217). The pilocytic astrocytoma represents a distinct subtype of glioma that is most common in children and is genetically and biologically distinct from the diffuse, infiltrating gliomas that affect both adults and children. It constitutes approximately two-thirds of all cerebellar astrocytomas and 28% of posterior fossa tumors (216). On gross inspection, the tumor arises most commonly from the cerebellum, where it frequently projects as a mural nodule into a macroscopically well-circumscribed fluid-filled cyst (Fig. 11.9). Solid (noncystic) variants also are encountered. Microscopically, the tumor is characterized by a “biphasic” pattern that consists of areas containing tightly packed, piloid (“hairlike”) tumor cell processes alternating with areas having numerous microscopic cysts. The latter often coalesce to form larger cysts. Nonspecific changes of astrocytic cell processes such as eosinophilic granular bodies and Rosenthal fibers also are characteristic. The latter are intracytoplasmic, opaque, homogeneous, strongly eosinophilic beaded masses that stain deep purple with Mallory’s phosphotungstic acid–hematoxylin. Their presence indicates a degenerative change, and they can be found in other tumors, notably in craniopharyngiomas. In an otherwise typical pilocytic astrocytoma, the presence of vascular proliferation
or nuclear atypia does not correlate with aggressive behavior as it does in diffuse astrocytomas. Mitoses are rare. Although grossly circumscribed, these tumors may locally infiltrate adjacent tissues and may extend into the adjacent subarachnoid space. Neither of these features suggests a tendency for aggressive behavior. Anaplastic (malignant) pilocytic astrocytomas are rare (218). Although most pilocytic astrocytomas have normal karyotypes (219), deletions on the long arm of chromosome 17 have been reported (220). A p53 mutation (exon 9, codon 324) was identified in one of seven pilocytic astrocytomas, but this was a silent mutation, and its significance is uncertain (221).
or nuclear atypia does not correlate with aggressive behavior as it does in diffuse astrocytomas. Mitoses are rare. Although grossly circumscribed, these tumors may locally infiltrate adjacent tissues and may extend into the adjacent subarachnoid space. Neither of these features suggests a tendency for aggressive behavior. Anaplastic (malignant) pilocytic astrocytomas are rare (218). Although most pilocytic astrocytomas have normal karyotypes (219), deletions on the long arm of chromosome 17 have been reported (220). A p53 mutation (exon 9, codon 324) was identified in one of seven pilocytic astrocytomas, but this was a silent mutation, and its significance is uncertain (221).
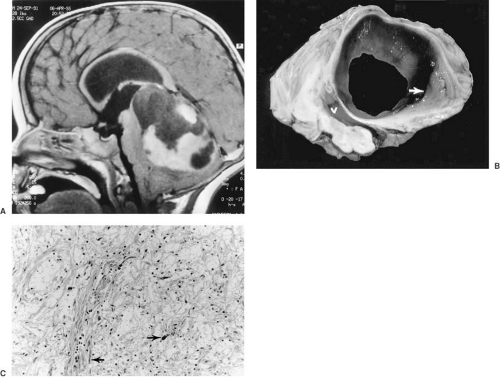 FIGURE 11.9. Cerebellar pilocytic astrocytoma consisting of a large, well-circumscribed tumor that compresses the fourth ventricle (v) (A and B). Arrow (B) indicates a mural nodule of solid tumor. C: The histologic section from the mural nodule of a pilocytic astrocytoma. The tumor contains abundant piloid (hairlike) processes and microscopic cysts. Arrows indicate Rosenthal fibers (hematoxylin and eosin, original magnification ×200). |
Diffuse astrocytomas can occur anywhere in the CNS and are as common in the posterior fossa as in the supratentorial space (222). Diffuse astrocytomas constitute a spectrum of infiltrating astroglial tumors that display increasing grades of anaplasia and include the low-grade diffuse (fibrillary) astrocytoma, anaplastic (intermediate grade) astrocytoma, and glioblastoma multiforme (high grade). Collectively, these tumors account for 12.4% of pediatric infratentorial tumors (216). These intra-axial tumors are similar in histology and biological behavior to those affecting adults (215,222,223). Common pathologic features of this group are extensive infiltration of CNS tissue and a tendency of lower grade lesions to progress to higher grade lesions. Diffuse (fibrillary) astrocytomas tend to arise in white matter and often extensively invade adjacent gray matter structures. In contrast to the pilocytic astrocytoma, these tumors are difficult to eradicate by surgery alone because of poor demarcation between tumor and surrounding normal tissues, and a higher biologic propensity for regrowth.
Histologically malignant astrocytomas (anaplastic astrocytoma and glioblastoma multiforme) of childhood are infiltrative, biologically aggressive neoplasms (Figs. 11.10 and 11.11) (224). Microscopically, they are characterized by perivascular pseudorosette formation, necrosis, high cell density or mitotic figures, and calcifications, which usually are minute. In this type of glioma, microcyst formation is less common (occurring in 23% of cases).
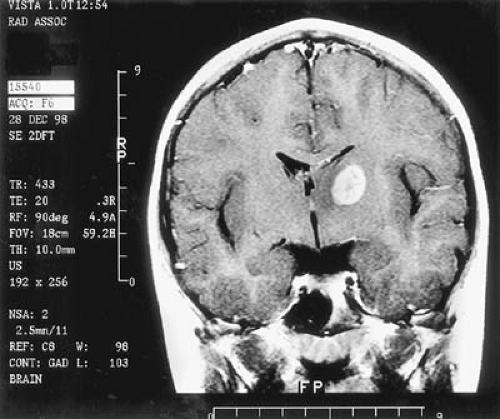 FIGURE 11.10. Coronal magnetic resonance imaging with contrast shows inhomogenously enhancing left thalamic anaplastic astrocytoma in a 9-year-old child with a 3-month history of right hand tremor. |
Cytogenetic characteristics of pediatric malignant astrocytomas differ from those most commonly encountered in adults. Structural abnormalities in chromosomes 9, 13, 17, and double minutes were observed in malignant astrocytomas (anaplastic astrocytoma and glioblastoma) of childhood, whereas the most common chromosomal abnormalities in adult glioblastomas are losses of chromosomes 10 and 19q or gains in chromosome 7 (219). Furthermore, to date, the chromosomal abnormalities found in malignant astrocytomas of childhood are similar to those reported in a genetically distinct subset of glioblastomas that was found to arise in young adults (225).
 FIGURE 11.11. Histologic section demonstrates increased cellularity and classic pseudopalisading necrosis of glioblastoma multiforme. |
Distinguishing between these various tumor types is difficult in many cases, and changes from one type of tumor to another have been noted with tumor recurrence (226). Additionally, spinal seeding with astrocytomas that appear benign on histologic examination has been observed (227).
Calcification of cerebellar tumors is usually minute but was seen in 26% of patients in Russell and Rubinstein’s series (215).
Primitive Neuroectodermal Tumor/Medulloblastoma.
The PNET/MB is the prototype of embryonal brain tumors and is among the most common malignant solid tumors of childhood. It accounts for 38% of pediatric infratentorial tumors (216). Unlike astrocytomas, MBs are derived from primitive neurons, the neuroblasts. Most of the laterally placed tumors arise from the fetal granular layer, a superficial layer of the cerebellar cortex present at birth, which disappears during the first year of life. The more numerous midline tumors are believed to arise from embryonal cell rests in the posterior medullary velum, the site of a germinal bud from which the external granular layer is derived (228). In some instances, the origin of the tumor has been traced back to the thirteenth week of gestation, a period of active cerebellar histiogenesis (229).
Macroscopically, these tumors are soft, friable, and moderately well demarcated from the remainder of the cerebellum. In general, they infiltrate the floor or lateral wall of the fourth ventricle and extend into its cavity (Fig. 11.12). The tumor grows backward, occluding the foramen magnum and infiltrating the meninges. It can metastasize by way of the CSF into the subarachnoid space of the spinal canal, with spinal (“drop”) metastasis characteristic. Less commonly, the tumor spreads over the cerebral convexity (Fig. 11.13). In the spinal canal, the cord
and cauda equina become coated with metastatic tumor deposits. The site most frequently involved is the lower end of the spinal cord.
and cauda equina become coated with metastatic tumor deposits. The site most frequently involved is the lower end of the spinal cord.
 FIGURE 11.12. Primitive neuroectodermal tumor/medulloblastoma invading the fourth ventricle and the midline cerebellum. |
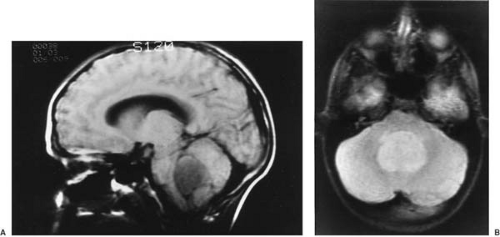 FIGURE 11.13. Primitive neuroectodermal tumor/medulloblastoma containing astrocytic elements. Sagittal (A) and axial (B) magnetic resonance images in this boy show a tumor within the fourth ventricle. It is of low signal intensity (darker than brain, but less dark than CSF) on the T1-weighted imaging (A). The lesion appeared to arise from the roof of the fourth ventricle. Although the neoplasm compressed the posterior aspect of the pons and medulla, it seemed separable from the brainstem. This was confirmed at surgery. (Courtesy of Drs. Hervey D. Segall and J. Gordon McComb, Departments of Radiology and Neurosurgery, Children’s Hospital, Los Angeles.) |
Histologically, PNET/MBs are highly cellular and are composed of small- to medium-sized cells with round to oval, often molded (carrot-shaped) hyperchromatic nuclei and poorly defined cell borders. Homer Wright rosettes (ring-like accumulations of tumor cell nuclei around a neurophil-containing or fibrillary core) are occasionally observed (Fig. 11.14). These tumors express a variety of neuronal/neuroendocrine markers, including synaptophysin (a 38-kd synaptic vesicle protein) and neurofilament proteins (230,231). Photoreceptor differentiation (retinal S-antigen and rhodopsin) is identified in 27% to 50% of these tumors (223,232). Glial fibrillary acidic protein (GFAP) has been reported in some 13% to 62% of PNET/MBs (233). One study found that patients with PNET/MBs that contained sheets or clumps of GFAP-immunoreactive neoplastic cells had a threefold increased risk of recurrence compared with those tumors without GFAP immunoreactivity. However, the prognostic value of immunohistochemical data has been controversial. Approximately 15% to 20% of children with PNET/MB have an anaplastic phenotype with a worse prognosis (234). In contrast, PNET/MBs with extreme nodularity have a more favorable prognosis (226,235). It also is important to know that posterior fossa PNET/MB and supratentorial PNET are molecularly distinct entities, although their histopathological analysis overlaps (236).
The most common molecular genetic abnormality identified to date in PNET/MB is loss of heterozygosity for the short arm of chromosome 17 [i(17q)], which occurs
in 30% to 50% of cases; more recent studies indicate that i(17q) is not an independent prognostic factor, but may be a marker for uncontrolled cell proliferation (237). Other common losses of heterozygosity involve 8p and 11p, 10p, 11q, 16q, 20q, and 20p. Gains have been found at 7q, 17q, 18q, 7p, 13q, 18p, and the like (238). Flow cytometric analyses have indicated a worse prognosis for DNA diploid tumors, whereas aneuploid tumors have a better prognosis (239,240). Several biological factors have been studied as candidate progonostic factors in PNET/MB: GFAP expression, calbindin-positivity, HER2/HER4 coexpression, MIB-1 proliferation index, mitotic index, TrkC mRNA expression and MYC mRNA expression (241,242). Of these, TrkC mRNA and MYC mRNA expression seem to be the most potent prognostic factors (243).
in 30% to 50% of cases; more recent studies indicate that i(17q) is not an independent prognostic factor, but may be a marker for uncontrolled cell proliferation (237). Other common losses of heterozygosity involve 8p and 11p, 10p, 11q, 16q, 20q, and 20p. Gains have been found at 7q, 17q, 18q, 7p, 13q, 18p, and the like (238). Flow cytometric analyses have indicated a worse prognosis for DNA diploid tumors, whereas aneuploid tumors have a better prognosis (239,240). Several biological factors have been studied as candidate progonostic factors in PNET/MB: GFAP expression, calbindin-positivity, HER2/HER4 coexpression, MIB-1 proliferation index, mitotic index, TrkC mRNA expression and MYC mRNA expression (241,242). Of these, TrkC mRNA and MYC mRNA expression seem to be the most potent prognostic factors (243).
 FIGURE 11.14. Histology of primitive neuroectodermal tumor of the cerebellum (primitive neuroectodermal tumor/medulloblastoma) with several Homer Wright rosettes (hematoxylin and eosin, original magnification ×1,000). |
The desmoplastic variant of PNET/MB is found in approximately 10% of PNET/MBs and tends to affect an older age group. In contrast to the classic midline form, it often arises laterally in a cerebellar hemisphere. The presence of reticulin-free “pale islands” is a characteristic histopathologic feature (244). Zacharoulis and colleagues have shown that desmoplastic pathology is more common in younger patients (median age 4.5 years for desmoplastic vs. 8.0 years for nondesmoplastic) and in the absence of other risk factors may be associated with a better prognosis, even for patients younger than three years of age (245).
Ependymomas.
Ependymomas are the third most common brain tumor after cerebellar astrocytoma and PNET/MB. They account for 6% to 15% of brain tumors in children (246). Although these tumors can arise from any part of the ventricular system, the roof and the floor of the fourth ventricle are the most common sites of origin in childhood (Fig. 11.15A). Grossly, fourth ventricular ependymomas are exophytic growths that arise from the ventricular floor and fill the ventricle. Tumor may extend into the basal subarachnoid space through the foramina of Lushka or protrude into the cisterna magna. Ependymomas invade adjacent parenchymal structures but are less overtly infiltrative than high-grade astrocytomas. In appearance, ependymomas are gray-tan, granular, and may contain cysts and areas of hemorrhage. Microscopically, the tumor is composed of a uniform population of cells with round to oval nuclei and fibrillary processes; the latter directed toward abundant intratumoral blood vessels forming a distinct perivascular pseudorosette pattern (Fig. 11.15B). Better differentiated examples may contain tubular structures and canals. Although ependymal tumors with increased cellularity, pleomorphism, mitoses, and necrosis have been termed anaplastic or malignant, such histological features have been less predictive of aggressive behavior than they are in diffuse astrocytomas (247). The association of a high number of mitoses and high cell density with incomplete pseudorosettes allows one to make the diagnosis of the anaplastic variant in supratentorial ependymomas (248). It is important to note that histologically benign ependymomas can hemorrhage and invade the brainstem (85). Paradoxically, the presence of calcifications in the tumor has a poor prognosis. Conversely, high cell density and nuclear polymorphism suggest a good prognosis (249). There is a paucity of histological markers in ependymoma that have strong predictive validity.
 FIGURE 11.15. A: Sagittal magnetic resonance imaging shows large contrast-enhancing ependymoma filling most of fourth ventricle. B: Histologic section of ependymoma shows a uniform population of cells with round to slightly oval nuclei and indistinct cell borders. Perivascular clear areas (pseudorosettes) represent tumor cell processes oriented perpendicular to intratumoral vessels (hematoxylin and eosin, original magnification ×500). |
Chromosomal abnormalities identified in ependymomas include monosomy and deletions of chromosome 22, trisomy of chromosome 7, loss of sex chromosomes, and structural rearrangements of chromosome 2 (223). In addition, chromosomes 9 and 11 may harbor genes involved in the pathogenesis of ependymoma. Integrative profiling of chromosome copy number abnormalities and gene expression is being developed to discriminate genetically between supratentorial and infratentorial ependymomas,
and between classical and anaplastic variants (250). A germ-line mutation of the tumor-suppressor gene p53 has been implicated in the pathogenesis of anaplastic ependymoma (251). Gaspar and colleagues have shown that the p53 pathway in ependymoma might be abrogated without p53 mutation, mdm2 overexpression, p14ARF deletion, or increased pax5 protein expression (54). A deletion of chromosome 1 has been identified in the myxopapillary ependymoma, a distinct subtype that arises in the filum terminale. Korshunov and colleagues examined gene expression patterns in relation to biologic behavior (252). Interestingly, spinal, supratentorial, and infratentorial ependymomas had different patterns of gene expression that could implicate a multistep transformation process leading to aggressive biologic behavior in some patients. It seems that the heterogeneity that applies at the chromosomal level also applies at the molecular level in ependymomas.
and between classical and anaplastic variants (250). A germ-line mutation of the tumor-suppressor gene p53 has been implicated in the pathogenesis of anaplastic ependymoma (251). Gaspar and colleagues have shown that the p53 pathway in ependymoma might be abrogated without p53 mutation, mdm2 overexpression, p14ARF deletion, or increased pax5 protein expression (54). A deletion of chromosome 1 has been identified in the myxopapillary ependymoma, a distinct subtype that arises in the filum terminale. Korshunov and colleagues examined gene expression patterns in relation to biologic behavior (252). Interestingly, spinal, supratentorial, and infratentorial ependymomas had different patterns of gene expression that could implicate a multistep transformation process leading to aggressive biologic behavior in some patients. It seems that the heterogeneity that applies at the chromosomal level also applies at the molecular level in ependymomas.
Other Tumors.
Other tumor types involving the cerebellum and its contiguous structures include undifferentiated sarcomas, some of which are of leptomeningeal origin, glioblastomas, and cerebellar hemangioblastoma, which is discussed in Chapter 12 under von Hippel–Lindau disease. Some of the uncommon CNS tumors are reviewed in Table 11.11 (253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


