Vigabatrin
Elinor Ben-Menachem
HISTORICAL BACKGROUND
Although it was known that decreases in brain gamma-aminobutyric acid (GABA) caused convulsions in animals and agents that enhanced GABA-mediated functions could have an anticonvulsant effect, the first suggestion that GABA might be an inhibitory neurotransmitter came from Elliot and van Gelder in 1958 (1).
Evidence for the anticonvulsant activity of GABA was gleaned from several observations: (a) focal agents that inhibit GABA synthesis, such as thiosemicarbazide and L-allylglycine, are proconvulsive; (b) focal agents that block the postsynaptic action of GABA, such as bicuculline and picrotoxin, are proconvulsive (2); (c) inhibition of glutamic acid decarboxylase (GAD) to block GABA synthesis causes seizures; (d) GABA receptor abnormalities are found in certain genetically determined epileptic syndromes in such animals as gerbils and audiogenic DBA/2 mice; (e) focal cortical infusion of GABA has a potent anticonvulsant effect; (f) drugs that enhance GABA-mediated inhibition are anticonvulsive in many animal models; (g) pyridoxine (i.e., the coenzyme of GAD) deficiency can cause seizures; and (h) pyridoxine or GABA itself can protect against seizures in vitamin B6-deficient children. These observations, as well as the recognition that GABA concentrations in cerebrospinal fluid (CSF) are decreased in some epileptic syndromes, supported the idea that diminished GABAergic inhibition contributes to seizure susceptibility.
Since the 1970s, several compounds have been developed that affect GABA subtype A (GABAA)-mediated inhibition at different levels and in different ways, depending on their mechanism of action. Vigabatrin (γ-vinyl-GABA) (VGB) is the only selective, irreversible GABA-transaminase (GABA-T) inhibitor that greatly increases whole-brain levels of GABA, presumably making it more available to its receptor site. It may also stimulate GABA release (3). VGB was synthesized in 1977, specifically as a substrate for GABA-T, by Jung and colleagues (4).
VGB is available worldwide, except in the United States, for use as an anticonvulsant. The agent has consistently been shown to be effective in the treatment of partial seizures and infantile spasms. The drug is not approved for use by the US Food and Drug Administration (FDA) because of the discovery of visual peripheral field defects occurring in what is likely more than 30% of patients (5).
GENERAL CHARACTERISTICS
VGB (4-amino-5-hexenoic acid, or γ-vinyl-GABA) is a structural analogue of GABA with a vinyl appendage (Fig. 66.1) rationally designed as an enzyme-activated irreversible, specific inhibitor of GABA-T (6,7). VGB is highly soluble in water, only slightly soluble in ethanol and methanol, and insoluble in hexane and toluene. This white to off-white crystalline solid has a melting point of 171°C to 177°C. The molecular mass is 129.16, and the conversion factor (CF) is 7.75 (mg/L × CF = μmol/L).
The drug exists as a racemic mixture of R(−) and S(+) isomers in equal proportions and exhibits no optical activity. Pharmacologic activity and toxic effects are associated only with the S(+) enantiomer; the R(σ) enantiomer is entirely inactive (8,9). No chiral inversion exists in humans. The major pharmacologic effects are determined by the half-life of the enzyme rather than by the drug or the S(+) enantiomer, because GABA-T, which is the target enzyme, has a much longer half-life than VGB itself (4,10).
MECHANISMS OF ACTION
Animal Studies
VGB causes very specific effects in the brain that are probably related to its anticonvulsant activity. The brain content of GABA, GABA-T, and GAD was measured over time after intraperitoneal injection of vigabatrin 1500 mg/kg into
mice. By 4 hours, whole-brain GABA had increased 5-fold, and GABA-T activity declined sharply. Recovery to 60% of baseline concentrations occurred after 5 days. A 30% decrease in GAD, demonstrated only at the high dose used, probably results from a feedback mechanism after the sudden increase in GABA concentration (4).
mice. By 4 hours, whole-brain GABA had increased 5-fold, and GABA-T activity declined sharply. Recovery to 60% of baseline concentrations occurred after 5 days. A 30% decrease in GAD, demonstrated only at the high dose used, probably results from a feedback mechanism after the sudden increase in GABA concentration (4).
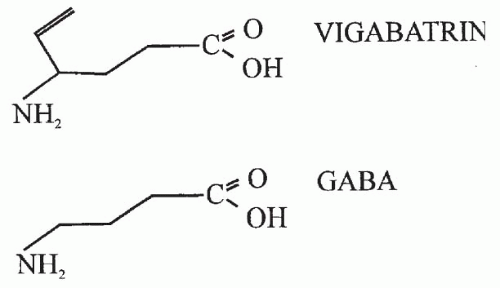 Figure 66.1 Chemical structure of vigabatrin. |
At high doses, VGB increases concentrations of β-alanine (an alternative substrate to GABA-T), homocarnosine (GABA and histidine), and hypotaurine while decreasing glutamine and threonine levels (11). Concentrations of free and total GABA and homocarnosine in the brain and CSF increase parallel to increasing doses of VGB (12).
Anticonvulsant Effects
VGB is inactive against maximal electroshock (MES)-induced seizures, bicuculline (i.e., GABA antagonist)-induced seizures, and pentylenetetrazol-induced seizures, unless injected directly into the midbrain of rats (13); however, an intravenous injection provided seizure protection after bicuculline-induced myoclonic activity (14), strychnine-induced tonic seizures (7), isoniazid-induced generalized seizures (7), audiogenic seizures in mice (15), photic-induced seizures in the baboon (16), and amygdala-kindled seizures in the rat (17).
Stereotaxic injections of small amounts of VGB into certain areas of rat brain provided seizure protection in relation to locally increased GABA levels (13). Seizure protection against MES was most prominent with local GABA increases in the midbrain tegmentum, including substantia nigra and midbrain reticular formation, but VGB injected into the thalamus, hippocampus, and cortex was not protective in this model. Seizure protection lasted up to 72 hours after a single injection to the substantia nigra. It was not until day 5 that the rats responded normally to MES. This finding supports observations that the rate of recovery of GABA-T is 5 days (4) and suggests that the anticonvulsant activity of VGB may be the result of the local increase in GABA levels rather than a direct effect of the drug.
Prevention of Epileptogenesis
Rats treated with saline or VGB were given repeated subconvulsive stimulation to the amygdala in an attempt to stimulate epileptogenesis (18). During 16 stimulations, saline-treated rats developed mild to fully developed generalized seizures, but VGB-treated animals remained seizure free. After a 10-day rest period, the rats previously treated with VGB progressed through the entire kindling procedure before developing fully kindled seizures, whereas saline-treated rats developed seizures immediately.
Neuroprotective Effects
In a Finnish study (19), rats pretreated with VGB were compared with rats pretreated with saline. Stimulation of the perforant pathway caused status epilepticus in all animals. On postmortem examination, the saline-treated rats had hippocampal cell damage and loss of somatostatincontaining interneurons in the dentate gyrus. VGB-treated rats, on the other hand, had histologically normal hippocampi. This finding suggests that VGB may have a neuroprotective effect on the hippocampus during status epilepticus (20).
Human Studies
Grove and associates (21) were the first to investigate the relationship between VGB and GABA in CSF as a means of determining the effects of this antiepileptic drug (AED) on brain neurotransmitters and amino acids. Patients with varied neurologic conditions were given VGB 0.5, 1, 2, or 6 g daily for 3 days. Free and total concentrations of GABA, β-alanine, homocarnosine, and VGB increased in a doseresponsive manner, except with 0.5 g per day, at which level no changes were noted. In another study (22), patients were treated with VGB 0.5 g twice daily, followed by VGB 1 g twice daily for 2 weeks and 2 weeks on placebo. At the end of the treatment, no changes from baseline were reported in homovanillic acid (HVA, the metabolite of dopamine) and 5-hydroxyindoleacetic acid (5-HIAA, the metabolite of serotonin), but dose-related increases were seen in free and total GABA and homocarnosine. At the end of the placebo period, GABA and homocarnosine levels had decreased to baseline values.
No consistent changes have been found in amino acids, HVA, or 5-HIAA in tissue and CSF with VGB 50 mg/kg taken for up to 3.5 years (23,24). In CSF, concentrations of acetylcholine, somatostatin, β-endorphins, prolactin, cyclic adenosine, or guanine monophosphate were also unchanged during long-term treatment (24,25). In a single-dose study, however, HVA and 5-HIAA concentrations increased initially up to 100%, but returned to baseline levels or slightly below after 1 month (10). At a 50-mg/kg dose, VGB caused a 200% to 300% increase in CSF and brain levels of GABA (26,27). A reduction in dose from 3 to 1.5 g per day proportionally decreased GABA levels in CSF (28). Dose and percentage increases in CSF GABA concentrations show a linear relationship. However, the relationship between dose increases and efficacy is more complex and may depend on the type of epilepsy. Kälviäinen and coworkers (29) suggested that responders to VGB monotherapy have higher glutamate levels (increased 14%) in the CSF before receiving
the drug than do nonresponders. Nuclear magnetic resonance spectroscopy in patients treated with VGB added to conventional AEDs confirmed results of GABA analysis in CSF (30,31); however, increased levels of glutamine and corresponding decreased levels of glutamate (by 9%) were identified, compared with levels in patients receiving conventional therapy alone.
the drug than do nonresponders. Nuclear magnetic resonance spectroscopy in patients treated with VGB added to conventional AEDs confirmed results of GABA analysis in CSF (30,31); however, increased levels of glutamine and corresponding decreased levels of glutamate (by 9%) were identified, compared with levels in patients receiving conventional therapy alone.
VGB also affects blood GABA and platelet GABA-T levels. At therapeutic doses of 2 to 3 g per day, platelet GABA-T is markedly reduced. A dose of 2 g per day maximally inhibits platelet GABA-T, with mean inhibition at approximately 70% (32). The concentration of plasma VGB is almost 10-fold that in CSF, and because platelets cannot regenerate GABA-T, the effect of VGB on this test system is also influenced by platelet regeneration.
ABSORPTION, DISTRIBUTION, AND METABOLISM
Absorption
Rate of Absorption
Peak VGB concentration is reached within 2 hours following administration of doses between 0.5 and 3 g (8,33,34). The absorption half-life ranges from 0.18 to 0.59 hours, and the mean terminal half-life is 5 to 7 hours. Approximately 60% to 80% of VGB can be recovered unchanged in a 24-hour urine collection, indicating a bioavailability of at least 60% to 80%. No metabolites are found in the urine, and the remaining amount of VGB probably disappears, as it is bound to GABA-T.
Distribution
Body Tissues and Fluids
The apparent volume of distribution of VGB is 0.8 L/kg (total body water is 0.6 L/kg) in volunteers, with the half-life of distribution at 1 to 2 hours. A comparison of the extrapolated (or initial) volume of distribution with the steady-state volume reveals that between 50% and 75% of VGB is outside the central blood compartment at steady state (22).
In one study of patients with epilepsy, the concentration of VGB in CSF was approximately 10% of blood levels (10). In this study, patients taking concomitant AEDs were given a single dose of VGB 50 mg/kg, and CSF and blood samples were taken for up to 5 days following the treatment. The highest VGB concentrations were found in CSF after the first sample. By 24 hours, only a trace was detectable in CSF, and no VGB was found at 72 hours or thereafter. Peak concentration in the blood was reached in 1 hour, decreasing thereafter to only small amounts detectable by 72 hours. The mean elimination half-life was 4.5 hours, and the AUC was 310 nmol/mL per hour. The VGB-CSF-to-blood ratio was 0.10. After a 3-year follow-up with doses of 50 mg/kg per day, CSF VGB levels were not significantly increased compared with the 6-month levels (23).
Protein Binding
Distribution in Placenta and Breast Milk
Passage of S(+) and R(−) VGB across the human placenta in vitro showed a low level of transfer from maternal to fetal blood and was comparable to that of other α-amino acids. Clearance for both enantiomers was about 27% of phenazone (38). The maximum amount of VGB that a nursing infant would ingest per day is approximately 3.6% of the R(−) and 1% of the S(+) enantiomer of the maternal dose of VGB (39).
Distribution in Children
Children demonstrate a lower AUC than adults, but renal clearance is the same in both populations. Children may need higher doses of VGB to achieve the same effect seen in adults.
Metabolism
Because no metabolites have been identified, it can be assumed that VGB is not metabolized in humans. Up to 82% of the oral dose is excreted unchanged in the urine (Aventis, data on file).
Elimination
Half-Life and Rate of Excretion
The elimination half-life of VGB is 5 to 8 hours, and the total clearance is about 1.7 to 1.9 mL/kg per min, with renal clearance accounting for 70% of the total. Elimination is not influenced by dose or duration of treatment (40). The biologic half-life is measured in days, not in hours.
Elimination in the Elderly
Renal and total-body clearances are slower in the elderly. Terminal half-life showed an inverse relationship to renal function, probably because of a smaller volume of distribution in patients with lower creatinine clearance (33). In patients with low renal clearance, extrarenal clearance may be compensatory. Reduced renal clearance causes an increase in the AUC-to-body-weight ratio. Patients with renal impairment, therefore, have higher plasma concentrations of VGB and a longer terminal half-life. The half-life in the elderly with reduced creatinine clearance is
approximately twice that in normal healthy volunteers. The AUC in elderly adults is 7-fold higher than in younger individuals because of reductions in renal clearance and volume of distribution. A nonlinear increase in the AUC-to-body-weight ratio occurs as creatinine clearance falls to < 60 mg per min.
approximately twice that in normal healthy volunteers. The AUC in elderly adults is 7-fold higher than in younger individuals because of reductions in renal clearance and volume of distribution. A nonlinear increase in the AUC-to-body-weight ratio occurs as creatinine clearance falls to < 60 mg per min.
EFFICACY
Adults
More than 18 years of clinical experience have confirmed the efficacy of VGB for patients with partial seizures and other specific seizure types.
Single-Blind Studies
Meta-analysis of the first 10 single-blind studies of VGB, enrolling 352 patients with highly intractable partial seizures, demonstrated that 55.8% of those treated had their seizure frequency reduced by more than 50% (22,28,41, 42, 43, 44, 45, 46, 47, 48). Another single-blind study (49) involved less severely intractable patients who had been experiencing four partial seizures per month, but were all socially integrated and active outpatients. Of these 19 patients, 14 (73%) had a seizure reduction greater than 50%, and 10 (53%) patients had a seizure reduction greater than 70%. Two patients developed new myoclonic jerks. No deleterious effects on cognitive function were noted.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


