Chapter 44 Wake-Promoting Medications
Basic Mechanisms and Pharmacology
Central Nervous Stimulants: Definitions
In this chapter, CNS stimulant is the generic term used for all wake-promoting compounds of potential use in the treatment of excessive daytime sleepiness (EDS). (See Chapters 4 and 45 for the classification of EDS disorders and the indications of CNS stimulants for patients affected with sleep disorders.) EDS is a common symptom in patients with sleep disorders and in the general population at large. CNS stimulants are generally effective on EDS independently of its underlying cause; however, they should be used cautiously because of their potential for misuse and abuse. In this chapter, we review the neurochemical, neurophysiologic, and neuropharmacologic properties of the CNS stimulants most commonly used in sleep medicine. This is followed by a perspective on future stimulant treatments.
Amphetamines and Amphetamine-Like Compounds
Historical Perspective
Narcolepsy was probably the first condition for which amphetamine was used clinically. It revolutionized therapy for the condition, although it was not curative. The piperazine derivative of amphetamine, methylphenidate, was introduced in 1959 by Yoss and Daly.1 The use of amphetamine in treatment of parkinsonism dates to 1937, when it was first used to alleviate muscle rigidity and postencephalitis parkinsonism. By 1968, its use in the treatment of this condition was largely suspended because of more effective dopaminergic agents. Until the dangers of amphetamine dependence and abuse became recognized, amphetamine was widely used in the treatment of obesity. It was also prescribed in the treatment of sedative abuse and alcoholism to offset sleepiness and lethargy.
Bradley and Bowen2 were the first to report on the use of amphetamine to modify antisocial behavior in children: “when children are withdrawn or lethargic, the amphetamine tended to make them more alert, more accessible to persons and the environment.” A paradoxical calming effect was also noted in some children and aggressive adults. Most notably, a selected group of children who were “hyperactive” tended to move more quietly, to be calmer, and to be less quarrelsome after being treated with amphetamine. In 1958, methylphenidate was introduced to treat hyperactivity in children (see reference 3). These observations preceded reports on the effects of amphetamine and methylphenidate in the hyperkinetic child (children diagnosed with what is now called attention-deficit/hyperactivity disorder [ADHD]).
Structure-Activity Relationships and Major Chemical Entities
Phenylisopropylamine (amphetamine) has a simple chemical structure resembling endogenous catecholamines (Fig. 44-1). This scaffold forms the template for a wide variety of pharmacologically active substances. Although amphetamine possesses strong central stimulant effects, minor modifications can result in agents with a broad spectrum of effects, including nasal decongestion, anorexia, vasoconstriction, and antidepressant effects (for the monoamine oxidase inhibitor tranylcypromine), and hallucinogenic properties (MDMA [methylenedioxymethamphetamine] and MDA [methylenedioxyamphetamine]).
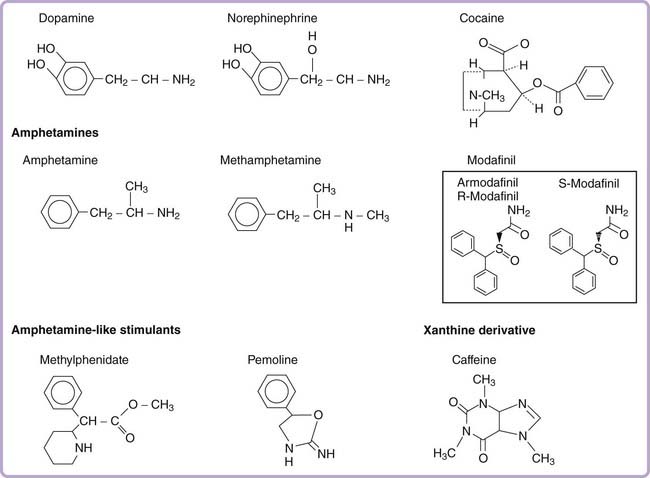
The phenylisopropylamine molecule can be divided into three structural components: (1) an aromatic nucleus, (2) a terminal amine, and (3) an isopropyl side chain. Substitution on the aromatic nucleus generally produces less potent if not entirely inactive stimulants.4 The substitution of two or more methoxy groups plus addition of ethyl, methyl, or bromine groups on the aromatic nucleus creates hallucinogens of various potencies. Ecstasy (MDMA) is built on a methamphetamine backbone, with a dimethoxy ring extending from the aromatic group. If a similar compound is synthesized with a primary amine (without the methyl group), it creates Love (MDA). Substitution at the amine group is the most common alteration. Methamphetamine, which is characterized by an additional methyl group attached to the amine (a secondary substituted amine), is more potent than amphetamine, probably because of increased CNS penetration. An intact isopropyl side chain appears to be needed to maintain stimulant efficacy. Changing the propyl to an ethyl side chain, for example, creates phenylethylamine, an endogenous neuroamine that has mood- and energy-enhancing properties but is less potent and has a much shorter half-life than amphetamine.
The pharmacologic effects of most amphetamine derivatives are isomer specific. These differential effects occur at both the pharmacokinetic level (absorption, brain penetration, metabolism, distribution volume, elimination) and the pharmacodynamic profile level (actual pharmacologic effects). D-Amphetamine, for example, is a far more potent stimulant than the L-derivative. In electroencephalographic (EEG) studies, D-amphetamine is four times more potent than L-amphetamine in inducing wakefulness.5 The relative effects of the D and L isomers of amphetamine on norepinephrine and dopamine transmission explain some of these pharmacodynamic differences (for details, see the pharmacology section for each compound). Not all effects are stereospecific, however. For example, both enantiomers are equipotent at suppressing rapid eye movement (REM) sleep in humans and rats and at producing amphetamine psychosis.
Amphetamine-like compounds, such as methylphenidate, pemoline, and fencamfamin, are structurally similar to amphetamines; all compounds include a benzene core with an ethylamine group side chain (see Fig. 44-1). Both methylphenidate and pemoline were commonly used for the treatment of EDS in narcolepsy, but pemoline has been withdrawn from the market in several countries because of liver toxicity (Table 44-1). The most commonly used commercially available form of methylphenidate is a racemic mixture of both a D and L enantiomer. In this preparation, the D-methylphenidate mainly contributes to its clinical effects, especially after oral administration. This is due to the fact that L-methylphenidate, but not D-methylphenidate, undergoes a significant first-pass metabolism (by deesterification to L-ritalinic acid). A single isomer form of D-methylphenidate is also marketed under the brand name of Focalin.
Table 44-1 Commonly Used Pharmacologic Compounds for Excessive Daytime Sleepiness
| STIMULANT COMPOUND (SCHEDULE) | USUAL DAILY DOSES* | T1/2 (hr); SIDE EFFECTS, NOTES |
|---|---|---|
| Amphetamine and Amphetamine-like CNS Stimulants | ||
| D-Amphetamine sulfate (II) | 5-60 mg (15 mg, 100 mg) | T1/2 16-30; Irritability, mood changes, headaches, palpitations, tremors, excessive sweating, insomnia |
| Methamphetamine HCl (II) | 5-60 mg (15 mg, 80 mg) | T1/2 9-15; Same as D-amphetamine; may have greater central over peripheral effects than D-amphetamine† |
| Methylphenidate HCl (II) | 10-60 mg (30 mg, 100 mg) | T1/2 3; Same as amphetamines; better therapeutic index than D-amphetamine, with less reduction of appetite or increase in blood pressure; short duration of action |
| Pemoline (IV) | 20-115 mg (37.5 mg, 150 mg) | T1/2 11-13; Less sympathomimetic effect, milder stimulant; slower onset of action, a tendency for drug buildup; may produce liver toxicity; had been with drawn from the U.S. market |
| Dopamine or Norepinephrine Uptake Inhibitor | ||
| Mazindol (IV) | 2-6 mg (NA) | T1/2 10-13; Weaker CNS stimulant effects; anorexia, dry mouth, irritability, headaches, gastrointestinal symptoms; reported to have less potential for abuse |
| Other Agents for Treatment of Excessive Daytime Sleepiness | ||
| Modafinil‡ (IV) | 100-400 mg (NA) | T1/2 9-14; No peripheral sympathomimetic action; headaches, nausea; reported to have less potential for abuse |
| Armodafinil (IV) | 150-250 mg | T1/2 10-15; Similar to those of modafinil |
| Monoamine Oxidase Inhibitor with Alerting Effect | ||
| Selegiline | 5-40 mg (NA) | T1/2 2; Low abuse potential; partial (10%-40%) interconversion to amphetamine |
| Xanthine Derivative | ||
| Caffeine§ | 100-200 mg (NA) | T1/2 3-7; Weak stimulant effect; 100 mg of caffeine is roughly equivalent to one cup of coffee; palpitations, hypertension |
CNS, central nervous system; NA, not applicable.
* Dosages recommended by the American Sleep Disorders Association are listed in parentheses (usual starting dose and maximal dose recommended).
† Methamphetamine is reported to have more central effects and may predispose more to amphetamine psychosis. The widespread misuse of methamphetamine has led to severe legal restriction on its manufacture, sale, and prescription in many countries. L-Amphetamine (dose range, 20 to 60 mg) is not available in the United States but probably has no advantage over D-amphetamine in the treatment of narcolepsy (slightly weaker stimulant).
‡ The half-life of s-enantiomer of modafinil is short at 3-4 hours, and thus the half-life of racemic modafinil mostly reflects the half-life of armodafini (r-enantiomer).
§ Caffeine can be bought without prescription in the form of tablets (NoDoz, 100 mg; Vivarin, 200 mg caffeine) and is used by many patients with narcolepsy before diagnosis.
Cocaine also mediates its psychostimulant effects by blocking catecholamine reuptake (mainly dopamine), but its structure is different from that of amphetamine-like compounds (see Fig. 44-1). The fact that cocaine and some dopamine transporter inhibitors are drugs of abuse is responsible for schedule labeling of such drugs by the Food and Drug Administration.
Molecular Targets of Amphetamine Action
The molecular targets mediating amphetamine-like stimulant effects are complex and vary according to the specific analogue or isomer used and the dose administered. Amphetamine increases catecholamine (dopamine and norepinephrine) release and inhibits reuptake from presynaptic terminals. This results in increase in catecholamine concentrations in the synaptic cleft and enhances postsynaptic stimulation. The presynaptic modulations by amphetamines are mediated by specific catecholamine transporters6 (Fig. 44-2). Axelrod and colleagues first demonstrated that epinephrine can be rapidly and selectively taken up by the heart, spleen, and glandular organs, each of which has significant sympathetic innervation. It was subsequently discovered that norepinephrine-containing neurons bind and take up norepinephrine against a concentration gradient, suggesting the existence of selective norepinephrine transporters. Further experiments also found that these transporters not only can carry catecholamine back into nerve terminals but also can release catecholamines by reverse efflux.
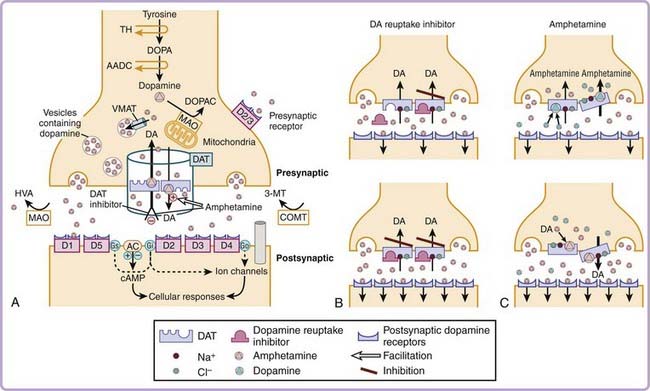
The molecules responsible, the dopamine transporter (DAT) and the norepinephrine transporter (NET), have now been cloned and characterized. The DAT and NET proteins are about 620–amino acid proteins with 12 putative membrane-spanning regions. Amphetamine derivatives are known to inhibit the uptake and to enhance the release of dopamine, norepinephrine, or both by interacting with the DAT and the NET. These transporters normally move dopamine and norepinephrine from the outside to the inside of the cell. This process is sodium dependent; sodium and chloride bind to the dopamine or norepinephrine transporter to immobilize it at the extracellular surface and to alter the conformation of the dopamine or norepinephrine binding site so that it facilitates substrate binding. Substrate binding allows movement of the carrier to the intracellular surface of the neuronal membrane, driven by sodium concentration gradients. Interestingly, in the presence of some drugs such as amphetamine, the direction of transport appears to be reversed (see Fig. 44-2). Dopamine and norepinephrine are thus moved from the inside of the cell to the outside through a mechanism called exchange diffusion, which occurs at low doses (1 to 5 mg/kg) of amphetamine. This mechanism, rather than a simple inhibition of monoamine reuptake, is involved in the enhancement of extracellular catecholamine release by amphetamine. It explains why amphetamine in particular is more potent than expected on the basis of its relatively low binding affinity for DAT and NET.7,8 A recent in vitro experiment has shown that amphetamine transport causes an inward sodium current. As intracellular sodium ions become more available, a DAT-mediated reverse transport of dopamine occurs, producing dopamine release through the DAT transporter.
At higher dose, other effects are involved. Increased serotonin (5-HT) release is also observed. Moderate to high doses of amphetamine (>5 mg/kg) also interact with the vascular monoamine transporter 2 (VMAT2; see reference 6). The vesicularization of the monoamines (dopamine, norepinephrine, serotonin, and histamine) in the CNS is dependent on VMAT2; VMAT2 regulates the size of the vesicular and cytosolic dopamine pools. Amphetamine is highly lipophilic and easily enters nerve terminals by diffusing across plasma membranes. Once inside, amphetamine depletes vesicular monoamine stores by several mechanisms. First, it binds directly, albeit with low affinity, to VMAT2, thereby inhibiting vesicular uptake. Second, amphetamine, a weak base, diffuses across the vesicular membrane in its uncharged (lipophilic) form and accumulates in the granules in its charged form (because of the lower pH of the synaptic vesicle interior). As vesicular amphetamine concentration increases, the buffering capacity of the catecholamine-containing vesicle is lost. The vesicular pH gradient diminishes, a loss of the free energy necessary for monoamine sequestration occurs, and vesicular monoamine uptake decreases. In addition, the collapse of the gradient purportedly results in a competition for protons between the native monoamines and amphetamine, thereby increasing uncharged vesicular neurotransmitter concentrations. All these mechanisms lead to a diffusion of the native monoamines out of the vesicles into the cytoplasm along a concentration gradient. Amphetamine can therefore be viewed as a physiologic VMAT2 antagonist that releases the vascular dopamine or norepinephrine loaded by VMAT2 into the cytoplasm. The high doses of amphetamine also inhibit monoamine oxidase activity. These mechanisms, as well as the reverse transport and the blocking of reuptake of dopamine or norepinephrine by amphetamine, lead to an increase in norepinephrine and dopamine synaptic concentrations (see reference 6), and these are independent on the phasic activity of the neurons.
Presynaptic Modulation of the Dopaminergic System Primarily Mediates the EEG Arousal Effects
How amphetamines and other stimulants increase EEG arousal has been explored by use of a canine model of the sleep disorder narcolepsy and DAT knockout mice models. Canine narcolepsy is a naturally occurring animal model of the human disorder.8 Similar to human patients, narcoleptic dogs are excessively sleepy (i.e., shorter sleep latency), have fragmented sleep patterns, and display cataplexy.8 Although amphetamine-like compounds are well known to stimulate catecholaminergic transmission, the exact mechanism by which they promote EEG arousal is still uncertain. Stimulation of either adrenergic or dopaminergic transmission or both has been suggested to play a role.
In narcoleptic and control Dobermans, the effects of ligands specific for the dopamine (GBR-12909, bupropion, and amineptine), the norepinephrine (nisoxetine and desipramine), or both the dopamine and norepinephrine (mazindol and nomifensine) transporters, as well as amphetamine and a nonamphetamine stimulant, modafinil, were studied to dissect wake-promoting mechanisms.9 The result indicated that prototypical dopamine uptake inhibitors, such as GBR-12909 and bupropion, dose dependently increased EEG arousal in narcoleptic dogs; nisoxetine and desipramine, two potent norepinephrine uptake inhibitors, had no effect on EEG arousal at doses that almost completely suppressed REM sleep and cataplexy (Fig. 44-3; see reference 9). Furthermore, the EEG arousal potency of various dopamine uptake inhibitors correlated tightly with in vitro DAT binding affinities (see Fig. 44-3), whereas a reduction in REM sleep correlated with in vitro NET binding affinities,9 suggesting that dopamine uptake inhibition is critical for the EEG arousal effects of these compounds.
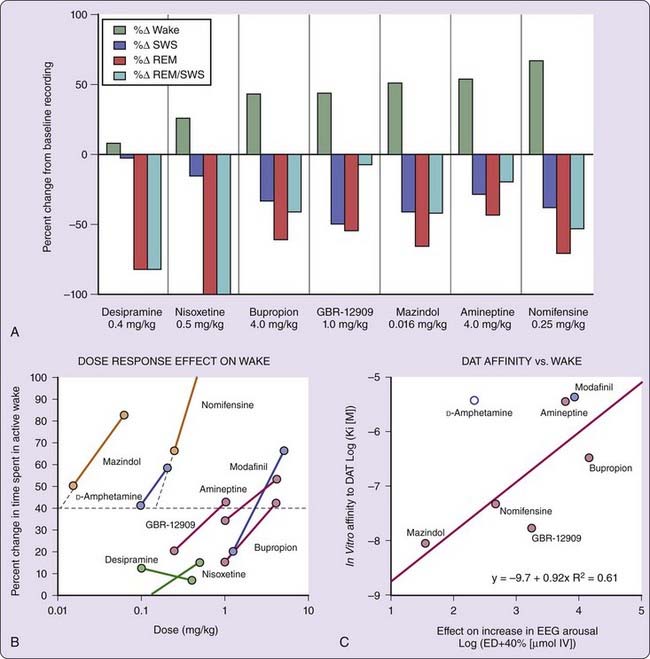
D-Amphetamine has a relatively low DAT binding affinity but potently (i.e., need for a low dose) promotes alertness (see Fig. 44-3). It is also generally considered more efficacious (i.e., can produce more alertness with high dose) than pure DAT reuptake inhibitors in promoting wakefulness. As described in the pharmacology section, D-amphetamine not only inhibits dopamine reuptake, it also enhances dopamine release (at lower dose by exchange diffusion and at higher dose by antagonistic action against VMAT2) and inhibits monoamine oxidation to prevent dopamine metabolism. The dopamine releasing effects of amphetamine are likely to explain the unusually high potency and efficacy of amphetamine in promoting EEG arousal.
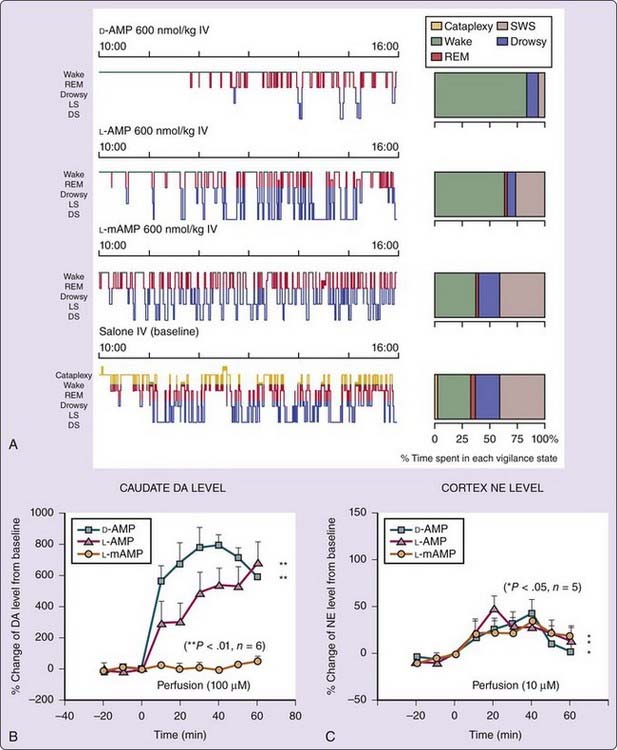
The effects of various amphetamine analogues (D-amphetamine, L-amphetamine, and L-methamphetamine) on EEG arousal and their in vivo effects on brain extracellular dopamine levels in narcoleptic dogs were compared10 to dissect wake-promoting effects of amphetamine. In vitro studies have demonstrated that the potency and selectivity for enhancing release or inhibiting uptake of dopamine and norepinephrine vary between amphetamine analogues and isomers.11 Amphetamine derivatives thus offer a unique opportunity to study the pharmacologic control of alertness in vivo. Hartmann and Cravens5 previously reported that D-amphetamine is four times more potent than L-amphetamine in inducing EEG arousal but that both enantiomers are equipotent at suppressing REM sleep in humans and rats. Enantiomer-specific effects have also been reported with methamphetamine; L-methamphetamine is much less potent as a stimulant than either D-methamphetamine or L– or D-amphetamine (see reference 11). Similarly, in canine narcolepsy, D-amphetamine is 3 times more potent than L-amphetamine and 12 times more potent than L-methamphetamine in increasing wakefulness and reducing slow-wave sleep (Fig. 44-4A).10
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


