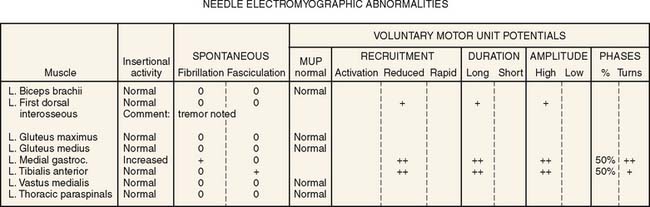
Figure 49-1 Values of attributes of nerve conduction of the patient.
The patient was asked to return to the electrophysiology laboratory for additional median motor studies so that the median motor terminal latency index (TLI) could be determined (Fig. 49-3). This calculated index provides a relative ratio of conduction slowing between the index finger and wrist, and elbow and wrist. The calculation is performed using basic motor studies and knowledge of the distance from wrist to thenar stimulating and recording sites [i.e., TLI = (1/DML) * (Dwrist-to-thenar/CV)] with distal motor latency (DML) and conduction velocity (CV). Using the data obtained from the tracing below, the calculated median TLI = 0.23 = (1/14.1 ms) *(70 mm/21 ms). Again dispersion was not seen, and the patient had no history of carpal tunnel syndrome or selective median sensory loss.
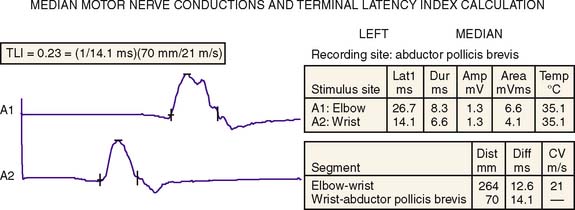
Figure 49-3 The patient’s median motor nerve conductions and terminal latency index (TLI) calculation.
Serum protein electrophoresis with immunofixation revealed an IgMκ monoclonal protein. Myelin-associated antibodies (MAG) by enzyme-linked immunosorbent assay and Western blot, as well as the cross-reactive sulfoglucuronyl paragloboside antibody (SGPG), were all positive [MAG = 1600: 51200; Western blot presence of 91–94 kDa band; SGPG = 3200: 204,800]. Genetic testing for commercially available HMSN1 was negative and included sequence and duplication analysis of peripheral myelin protein 22 and sequencing of myelin protein zero, and connexin 32. The following laboratory tests were normal: hemoglobin, electrolytes, plasma glucose, serum thyroid stimulating hormone, B12, and folate. Excessive alcohol use was denied.
The patient underwent bone marrow biopsy without identified lymphoplasmacytic lymphoma (Waldenström’s syndrome) or amyloidosis. Treatment with rituximab at 375 mg/m2 given as an intravenous (IV) infusion once weekly for four doses. Improvement was observed in proprioception and gait without improvement in tremor.
CONCLUSION
Monoclonal gammopathies (paraproteins or monoclonal proteins) are associated with a wide range of peripheral neurologic disorders. These proteins are produced by proliferating clones of plasma cells of different immunoglobulin types (IgM, IgG, IgA). Infiltrative and remote immune effects of these plasma cell disorders have been described. Among those with nerve infiltration is AL amyloidosis and intraneural lymphomas. However, parainflammatory effects of these plasma cell disorders are likely more common. One robust but rare example of such a remote effect is the syndrome collectively termed POEMS for polyneuropathy organomeglia endocrinopathy monoclonal protein and skin changes. Multiple elevated immune markers affect these different tissues, including nerve. More common, however, is the disorder presented here, namely a neuropathy associated with IgM monoclonal proteins and also judged to be sufficiently characteristic to justify the use of the acronym distal acquired demyelinating symmetric (DADS) neuropathy. Antibodies directed against MAG (anti-MAG) may explain the clinical, electrophysiologic, and histologic features of the DADS neuropathy phenotype.2 Patients present with insidious chronic sensory ataxia. Some studies, however, have found little or no differences (and not statistically significant) between the type and severity of neuropathies in patients with or without anti-MAG antibodies.3,4 Low levels of these antibodies in many IgM individuals without neuropathy, and/or alternative pathogenesis, for example, IgM amyloidosis, may also argue against their definitive role in pathogenesis.5 Nevertheless, among those with highest titers of MAG antibodies and with Western blot positivity, the association with neuropathy is made and was supportive of the patient’s diagnosis. Specifically, our patient had very high MAG antibody positivity and additional positivity with the cross-reactive SGPG.
The presented patient was initially believed to have HMSN1 when a chronic course, absence of positive sensory symptoms (i.e., no prickling, tingling, or paresthesias), and nerve conductions showed demyelination without dispersion. These clinical and electrophysiologic features are salient to HMSN1.6 Furthermore, postural tremor in HMSN1 were described pre- and postgenetic discoveries and such HMSN1 patients with tremor have sometimes been designated with Roussy-Lévy syndrome after the original descriptors.7 Tremor and the other features described in this patient are also common in IgM-associated neuropathy.8 At present, however, using knowledge of the distal predominant demyelination associated with DADS, a distinguishing nerve conduction study is potentially able to distinguish HMSN1 from this IgM-associated neuropathy.9 Specifically, the TLI = (1/DML [milliseconds] × (distal conduction distance [millimeters]/distal MCV [meters/second]) of the median nerve less than 0.26 was able to distinguish HMSN1 from IgM MAG-associated neuropathy in most cases, and was helpful in the presented case.
Treatment of the monoclonal neuropathies has historically been problematic with IgM-associated neuropathies, which are often refractory. For those without a major deficit, supportive measures of gait safety (i.e., using a cane, good footware, and safety) may be best in many with ongoing surveillance of the plasma cell disorder. For our patient, the worsening of condition threatened his employment; also, his safety was threatened by frequent falls. His treatment was initiated on rituximab after several case series suggested possible benefit.10–12 In our patient, the treatment did help his neuropathy, preventing falls and allowing him to remain employed. It did not help his tremor, which remains problematic.
Related posts:
 An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
An Unusual Case of Pia-Arachnoiditis due to Metastatic Breast Carcinoma
 Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
Stepwise Facial and Upper Limb Weakness and Small Fiber Sensory Loss—Tangier Disease
 Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
Late-Onset Transthyretin Val30Met Familial Amyloid Polyneuropathy Unrelated to Endemic Foci
 Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
Inflammatory Multifocal Encephalomyelopolyradiculoneuropathy Mimicking a Granulomatous or Lymphomatous Process
 Chronic Inflammatory Demyelinating Polyradiculoneuropathy–Like Neuropathy with Gastrointestinal Dysmotility, Ophthalmoplegia, and Leukoencephalopathy—Mitochondrial Neurogastrointestinal Encephalopathy
Chronic Inflammatory Demyelinating Polyradiculoneuropathy–Like Neuropathy with Gastrointestinal Dysmotility, Ophthalmoplegia, and Leukoencephalopathy—Mitochondrial Neurogastrointestinal Encephalopathy
 Amyloidosis Typing Based on Laser Microdissection and Mass Spectrometry of Paraffin-Embedded Tissue Biopsies
Amyloidosis Typing Based on Laser Microdissection and Mass Spectrometry of Paraffin-Embedded Tissue Biopsies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


