Sensory deficit
Values are expressed as the mean ± SD.
ADL, activities of daily living.
* Based on previous reports.1,4
In contrast to conventional FAP ATTR Val30Met with a relatively early age at onset in the endemic foci, we have reported patients carrying the same mutation but with a late age of onset (50 years or older) and no relationship to the endemic foci.6 The typical features of these cases are distinct from those related to the endemic foci. These differences include a low penetrance rate; relatively mild autonomic dysfunction; loss of all sensory modalities rather than sensory dissociation; the frequent presence of cardiomegaly; extreme male preponderance; and an absence of anticipation of the age at onset (see Table 30-1).1–4,6 Before the availability of molecular diagnosis, this type of FAP ATTR Val30Met had been overlooked because its clinical features differ from those of the “typical” early-onset cases in the endemic foci. However, late-onset FAP ATTR Val30Met is now considered to be more prevalent in Japan and elsewhere than was previously believed, but it is still not widely recognized.3 Wide geographic distribution, sporadic occurrence, and atypical clinical features may still result in missed diagnoses unless pathologic confirmation of amyloid deposition or molecular diagnosis is actively pursued.
In the present study, we describe a patient with late-onset FAP ATTR Val30Met unrelated to the endemic foci to further characterize this type of FAP with apparently nonspecific clinicopathologic features.
CASE REPORT
A 72-year-old man noted numbness in the upper and lower extremities 3 years before admission to our hospital. Over the next 2 years, the numbness gradually progressed in a centripetal pattern, and muscular weakness developed in the distal portions of the upper and lower extremities. However, there were no symptoms related to autonomic nerve dysfunction. He was admitted to a hospital, where a sural nerve biopsy was performed. The biopsy specimen showed severe myelinated fiber loss, in which large and small myelinated fibers were decreased to a similar extent. Congo red staining of the biopsy sample revealed no amyloid deposition. Intravenous immunoglobulin therapy was given based on a tentative diagnosis of immune-mediated neuropathy, but no effect was noted. Since then, the patient has been followed up as a neuropathy of unknown etiology. At age 75, he was referred to our hospital. Although diarrhea became frequent at that time, other symptoms related to autonomic dysfunction were not reported. His mental state was good. The cranial nerves were intact. There were no vitreous opacities. Loss of light touch, position, vibration, and pain sensation was present in the leg distal to the knee and the hands. Muscular weakness occurred in a moderate to severe degree in the distal portions of the four extremities. Muscular atrophy was also obvious in the hands and legs. Steppage gait was obvious and he could barely walk by himself. Deep tendon reflexes were absent in the upper and lower extremities. Plantar responses were flexor on both sides.
Blood cell counts, blood chemistry, and urine analysis showed no appreciable abnormalities. Cerebrospinal fluid examination revealed a slightly elevated protein level (53 mg/dL) with no abnormality in cell count. Motor conduction velocity, distal latency, and amplitude of compound muscle action potential in the median nerve were 22 m/s, 9.4 ms, and 0.3 mV, respectively. Compound muscle action potential in the tibial nerve and sensory nerve action potentials in the median and sural nerves could not be elicited. Electrocardiogram showed reduced variation in R-R intervals but no conduction block. Echocardiographic examination revealed a thickening of the ventricular wall with a hyperechoic area, compatible with cardiac amyloidosis. Blood pressure was 126/79 (systolic/diastolic) mm Hg in the supine position and 82/47 while standing. Cardiac (123) I-meta-iodobenzylguanidine (MIBG) uptake confirmed a reduction of sympathetic nerve activity: the mean 123I-MIBG heart/mediastinum ratio was 1.33 in the early scan and 1.15 in the delayed scan.
Although family history of peripheral neuropathy or amyloidosis was not reported, the presence of autonomic dysfunctions leads us to consider the diagnosis of FAP. A heterozygous Val30Met mutation in the transthyretin gene was identified by DNA analysis.
CONCLUSIONS
Through the development of gene diagnostic techniques, transthyretin-related FAP has been shown to exist in many nations worldwide. For example, sporadic late-onset FAP ATTR Val30Met is apparently not as rare in Japan as previously thought.5 However, the classic concept of FAP is strong in the minds of many neurologists, which often results in a delay in the correct diagnosis of sporadic cases.5 Thus, recognizing the variability in the clinical, electrophysiologic, and histopathologic features of FAP, especially for sporadic late-onset FAP ATTR Val30Met in nonendemic areas, is becoming more important.
Marked autonomic dysfunction and the presence of sensory dissociation reflected by predominant small fiber loss including unmyelinated fibers have been considered to be the characteristic features for a textbook example of polyneuropathy associated with amyloidosis, including FAP ATTR Val30Met (Fig. 30-1A).7,8 On the contrary, these features are not common in late-onset FAP ATTR Val30Met in nonendemic areas (see Fig. 30-1B).1 Neuropathic features in this form of FAP may be nondescriptive, showing both superficial and deep sensory impairment without obvious autonomic symptoms, especially in the initial phase. Indeed, our experience suggests that physicians may not consider FAP until amyloid deposition is detected in sural nerve biopsy specimens. However because the amount of amyloid deposition is less in this form of FAP as compared with the conventional early-onset form from endemic foci,2 amyloid deposition may appear negative with Congo red staining as we observed in our case.6 Therefore, the possibility of sporadic late-onset FAP ATTR Val30Met should be kept in mind while examining patients with late-onset neuropathy of undetermined etiology. Careful examination for autonomic symptoms and cardiac amyloid deposition may help determine a correct diagnosis.
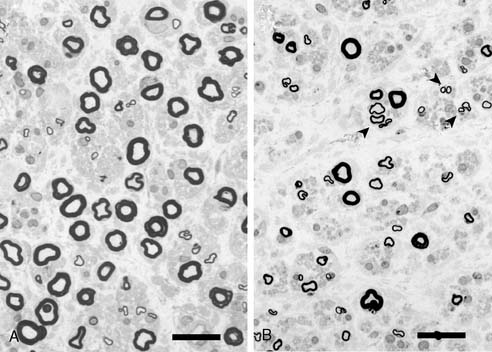
Figure 30-1 Representative photographs of sural nerve biopsy specimens from an early-onset case from the endemic foci (A) and a late-onset case from a non-endemic area (B). A, Small myelinated fibers show more loss than large myelinated fibers. B, Predominant small fiber loss as in A is not observed. Axonal sprouting is present (arrowheads). Bar = 20μm.
Related posts:
 Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Focal Mononeuropathy Onset of Amyotrophic Lateral Sclerosis
A Case of Guillain-Barré Syndrome Associated with Anti-GD1b Immunoglobulin G Antibodies
Length-Related Axonal Loss in Neuropathy
Malignant Peripheral Nerve Sheath Tumor
Disseminated Sporotrichosis with Multiple Granulomatous Mononeuropathies
Late Sporadic CMT4C—A New KIAA1985 Mutation
Focal Mononeuropathy Onset of Amyotrophic Lateral Sclerosis
A Case of Guillain-Barré Syndrome Associated with Anti-GD1b Immunoglobulin G Antibodies
Length-Related Axonal Loss in Neuropathy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


