5 Ancillary xamination
Laboratory Findings
Leon Sazbon
Although some laboratory parameters have prognostic significance in acute head injury, they are of minimal or no value for evaluating the vegetative state (VS). In patients in VS, laboratory tests are used mainly as a prophylactic screen to detect and follow up complications.
In acute coma, stress-induced acute hyperglycemia, which is often accompanied by impaired oxidative metabolism, leads to high serum lactate values and metabolic acidosis [1,2]. These alterations are of short duration and not generally seen in VS. If they persist, they may be predictive of irreversible VS [2].
Hypoglycemia, serum or urine electrolyte imbalance, leukocytosis, and accelerated sedimentation rate are all frequent findings related to multiple complications.
The development of periarticular bone formation may increase alkaline phosphatase levels [3]. Occult blood in feces or gastric juice is almost always present, with gastric acidity and normal pH [4]. Hypoproteinuria and inversion albuminoglobulins are linked to nutrition or hepatic disorders (see p. 19, 22).
Changes in hormone profile are described in the section on medical complications in Chapter 4 (see p. 23).
The first stage of acute injury is associated with enzymatic changes in serum or cerebrospinal fluid, or both: creatine phosphokinase (CPK) and its isoenzymes, mainly CPK1 of the brain type in the brain (also known as CKBB); lactic dehydrogenase (LDH) and its isoenzymes; and glutamic oxalacetic transaminase (GOT) and pyruvates. Levels of all of these generally return to normal in the stage of VS [2,5–8].
Low cerebrospinal fluid cAMP was observed by Rudman et al. [9] and Sazbon [10]. A trend toward normalization of this parameter might indicate a good prognosis.
A certain degree of hypoxia, with PO2 values of 60–80 mmHg in arterial blood, is the rule. Recovered patients show a marked diminution of forced vital capacity and both expiratory and inspiratory reserves [11]. This may be explained by paresis of the respiratory muscles (except the diaphragm), or the effect of possible bronchial aspiration, with subsequent inflammation of the pulmonary parenchyma by irritative acid gastric contents, alveolar collapse, microatelectasis, and interstitial sclerosis.
The increase in adrenocortical parameters – namely cortisol, norepinephrine, and catecholamines – which is characteristic of the first days after brain injury, regresses and then almost completely disappears in VS [5,12,13].
Pezzilli et al. [14] reported that hyperamylasemia and, less frequently, hyperlipidemia occur in the first phase of unconsciousness, but without proof of pancreatic involvement. These observations require further study.
A decrease in serum zinc concentrations, with loss of urinary zinc, is the rule in acute coma and is clearly correlated with the extent of brain damage. These parameters usually normalize within 2 weeks, although they have rarely been observed in VS [15,16]. The cause of the hypozincemia may be stress [17].
In the last few years, genetic factors such as the ε4 (epsilon 4) allele of apolipoprotein E have been found to have possible prognostic significance in post-traumatic coma and VS. Patients with this gene were likely to have a bad outcome [18–20].
Neuroimaging in the Vegetative State
Hans Hacker
The pathological conditions underlying the clinical picture in patients in the vegetative state are well known. Important information concerning the anatomical and pathological state of the brain [21,22], its functional, vascular, and chemical state, and regional cerebral blood flow, can be obtained using computed tomography (CT) [23], magnetic resonance imaging (MRI) [24,25], functional MRI, magnetic resonance angiography (MRA), MRI perfusion studies, magnetic resonance spectroscopy, and positron emission tomography (PET) [26]. In the usual clinical surroundings, many of these sophisticated techniques are not available. The absence of cooperation of patients in a vegetative state excludes complicated or long examinations without general anesthesia. It is also debatable how much of the information that can be obtained is relevant to the treatment or prognosis. At present, only a detailed anatomical diagnosis of brain lesions and vascular efficiency is indispensable. This is best obtained with MRI and MRA or, if these are not possible, with CT [27] contrast imaging.
The imaging findings at the time of examination, which may well be weeks after the injury, may conceal the extent and multiplicity of brain lesions in the immediate post-traumatic phase. For this reason, it is necessary to collect all previous examinations as well, although this is sometimes difficult to achieve. Reading and interpreting a series of older examinations, with a large number of films, and comparing them with the current examination is always a time-consuming and difficult task, but it is indispensable. Finally, a detailed description of the development of each of the different lesions over the previous few weeks should be presented.
Many types of traumatic brain lesion are demonstrated by imaging techniques:
• Compression by external hematomas: epidural; subdural, acute or chronic
• Brain swelling with internal shifting of parts of the brain, herniation, and compression of vessels
• Open or penetrating brain lesions
• Cortical lacerations
• Contusions
• Hemorrhages
• Subarachnoid bleeding
• Infarcts
• Shearing injuries
• Diffuse axonal injuries (DAIs)
• Vascular occlusions (neck or brain)
• Hydrocephalus: acute or low pressure
• Secondary lesions: thrombosis, infection
There is agreement among neuropathological and neuroradiological investigators that the most frequently found lesions in VS are:
• Diffuse axonal injuries (DAIs)
• Shearing injuries
• Extended cortical contusions
The most frequent locations for DAIs are:
• Corpus callosum and corona radiata
• Dorsolateral brain stem
Less frequent locations include:
• Bilateral thalamus
• Parahippocampal gyrus
• Ventral brain stem and medulla oblongata
The following grading of DAIs and shearing injuries is used:
• Grade 1: hemispheric white matter
• Grade 2: hemispheric white matter and corpus callosum
• Grade 3: hemispheric white matter, corpus callosum, and brain stem
A careful statistical study of lesions in 80 patients in post-traumatic vegetative state presented by Kampfl et al. showed that lesions in the corpus callosum and dorsolateral upper brainstem correlate with failure to recover [28,29]; this would correspond to grade 3. Most patients present with multiple locations of lesions with different pathologies. There have been no reports of patients with a single lesion or no lesions on MRI [30]. The main location observed in more than 40% of patients by Kampfl et al., are:
• Lobar white matter, mainly in the frontal and temporal lobes
• Cortical contusions
• Corpus callosum
• Thalamic and basal ganglia
• Parahippocampal region
• Dorsolateral brain stem
• Rostral ventral brain stem and medulla oblongata
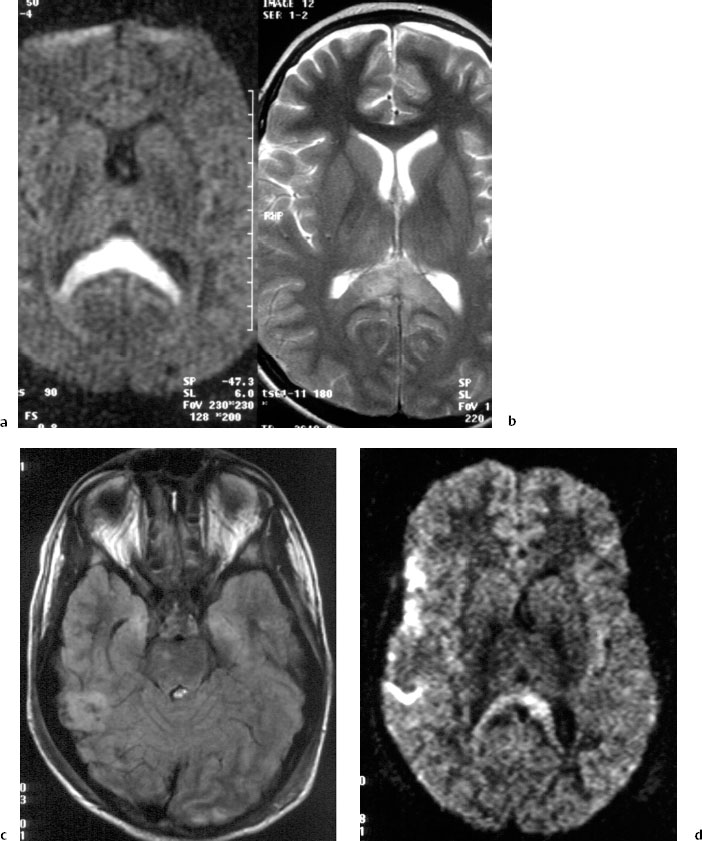
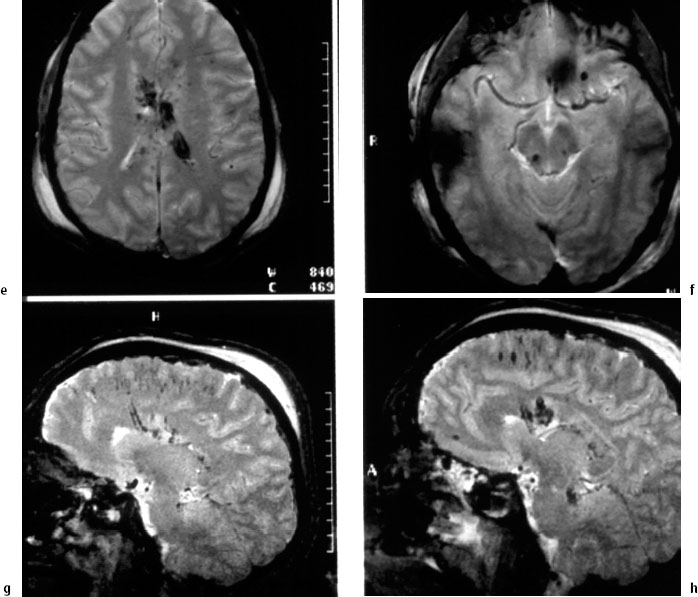
Fig 5.1 Case 1: a 12-year-old child with coma following a fall from horseback. Magnetic resonance imaging (MRI), 2 days after the trauma.
a Diffusion-weighted imaging (DWI).
b Spin-echo T2-weighted MRI, showing a lesion in the posterior corpus callosum, which is more clearly evident on the diffusion-weighted image.
c Fluid-attenuated inversion recovery (FLAIR). There are cortical and subcortical lesions in the right temporal lobe and in the medial parts of both temporal lobesparahippocampal gyrus, right pons.
d DWI, with an axial section higher than in c. Lesions are seen in the right temporal lobe, right operculum, and right and left cingulum.
e—h Gadolinium-enhanced T2*-weighted MRI. There are lesions in the corpus callosum, multiple diffuse axonal injuries with subcortical microhemorrhage, in the corona radiata, and in the right lateral dorsal mesencephalon
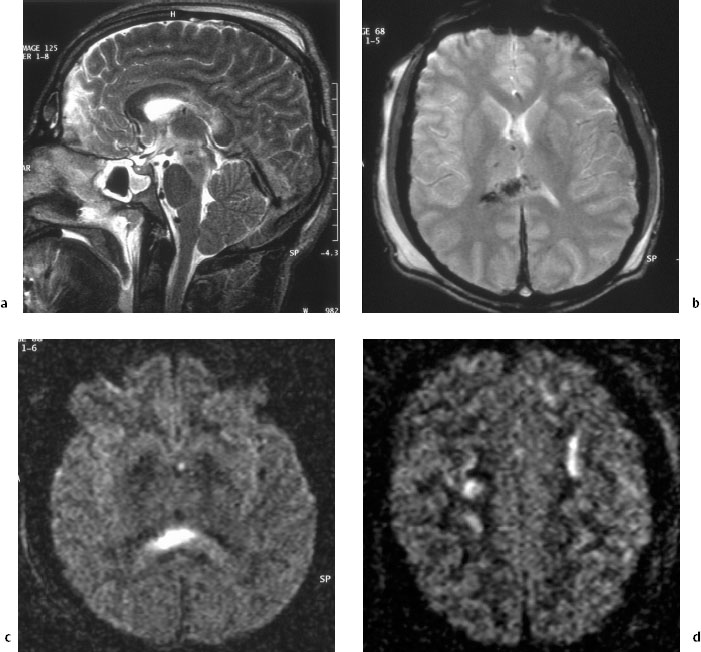
Fig 5.2 Case 2: a 31-year-old man who initially had coma after a car accident, with a vegetative state after 6 weeks. Magnetic resonance imaging (MRI) 6 weeks after the trauma.
a Spin-echo T2-weighted MRI. Lesions include frontal cortical defects, destruction and atrophy of the posterior part of the corpus callosum, and diffuse axonal injury in the rostral mesencephalon.
b Gadolinium-enhanced T2-weighted MRI. There are frontal cortical contusions, hemosiderin in the right thalamus, and lesions in the basal ganglia and corpus callosum.
c Diffusion-weighted imaging (DWI). There is a lesion in the corpus callosum.
d DWI. Diffuse axonal injury in the centrum semiovale
Lesions interfering with the connection between central and cortical structures, in addition to partial interruptions of transcallosal connections, are likely to be at the origin of locking-out from perception and reaction to external stimuli. These lesions are best seen during the first few weeks. Lesions found in the basal ganglia, especially when bilateral, also contribute to loss of consciousness. If brain-stem lesions are visualized, an impaired arousal reaction is explained. Pathological MRI findings in two patients are shown in Figs. 5.1 and 5.2.
Evidence in perfusion studies of a reduced regional cortical blood flow adds to the understanding of coma. It can also be assumed that a persistent flow rate below 20 mL/100 g/min provokes cortical atrophy. The value of spectroscopic magnetic resonance studies [31] is as yet unclear. In the future, spectroscopic imaging may allow the detection of regions in which there is no recovery. At present, CT studies in the acute stage and MRI in the postacute phase are the crucial examinations.
MRI examinations should include the following sequences:
• Spin-echo (SE) T1-weighted
• Proton density
• SE T2-weighted
• Gadolinium-enhanced (GE) T2*
These can be supplemented by the following techniques, if available:
• Fluid-attenuated inversion recovery (FLAIR)
• Diffusion-weighted imaging (DWI)
And, if vascular occlusion is suspected:
• Magnetic resonance angiography (MRA)
T2* images are sensitive for hemosiderin, and even after many weeks may indicate the presence of tiny shearing injuries. FLAIR and DWI are superior for demonstrating small and faint shifts in tissue water and of small foci of demyelination, allowing visualization of small DAIs.
Check-up examinations to assess the development of hydrocephalus can be carried out using CT, which should be performed every 3 months, or more frequently, during the first post-traumatic year.
Recent publications [32] on magnetic resonance spectrography in the mesencephalon have reported that reduced nuclear aspartic acid (NAA) appears to be predictive of a poor outcome. Confirmation of this finding by other groups must be awaited.
Acknowledgment
I am grateful Dr. S. Weidauer and Prof. Zanella at the Institute of Neuroradiology, University of Frankfurt, Germany, for kindly providing the clinical cases.
Neurophysiological Assessment of Brain Function in the Persistent Vegetative State
Alessandro Pincherle, Walter G. Sannita
The electrophysiological signals recorded by macroelectrodes from the surface of scalp (or, in selected instances, brain cortex) reflect spatially averaged postsynaptic potentials originating in large neuronal populations, notably ionic currents generated by membrane and volume-conducted through the extracellular media [34–36]. The functions of cerebral cortex are implicitly accepted to depend to a significant extent on the integrated activity of neuronal assemblies and circuitry. The hypothesis that collective signals recorded by macroelectrodes reflect with acceptable approximation the activity of these neural populations is therefore practicable. Experimental evidence and clinical observations converge in suggesting some functional correlation between neuronal mechanisms of, for example, sensory information processing, higher (“cognitive”) performance, sleep-wakefulness cycle, vigilance, etc., and the electrophysiological signals generated in the activated neuronal assemblies. This correlation allows some extrapolation from single-unit/multi-unit cell recording to human data and privileged methods of investigation [37,38]
Electroencephalography
Spontaneous brain signals are recorded routinely for diagnostic purposes using electroencephalography (EEG) [39]. Under properly controlled experimental conditions (and with due approximation), the technique has also proved a reliable method in brain research, as it is noninvasive and can be applied repeatedly with minimal discomfort even in the absence of collaboration by the patient. Remarkably, the EEG is modulated by processes that also regulate vigilance [40]. These peculiarities make it a selective approach to the investigation of brain function or malfunctioning in intensive-care routine and, eventually, in research on the electrophysiological concomitants of vigilance and consciousness [41]. EEG transient or rhythmic patterns (e. g., triphasic complexes in hepatic encephalopathies, asymmetric or focal slow activities in supratentorial hemispheric lesions, diffuse EEG slowing, etc.) provide the clinician with indications of diagnostic relevance also in the absence of clinical signs [42]. Epileptic events without motor symptoms can be documented in comatose patients only by EEG recordings. The sleep phases are classified on the basis of EEG patterns (slow waves, spindles, sharp waves, K complex, etc.) that also provide information about the responsiveness of the thalamocortical system and its abnormalities. A variety of EEG patterns have been observed in patients in coma or vegetative state, including burst-suppression, bilateral periodic complexes, epileptic discharges, diffuse nonreactive alpha activity, spindle activity, and other abnormalities with or without pathological connotation.
Dolce and Kaemmerer [43] studied the EEG evolution from onset of coma to outcome in five patients. Three patients died, one recovered completely, and one only partially; the duration of coma was 2–5 weeks, with a subsequent vegetative state lasting 4–40 weeks. The EEG recordings showed slow (delta) waves with low amplitude and no detectable reactivity. During the evolution into a vegetative state, the EEG became progressively rhythmic in the 4–6 Hz frequency range and slightly reactive. In the late recordings, a 6–7 Hz activity was observed, with the delta activity (if present) restricted to the scalp locations over the lesional areas. The longitudinal EEG and clinical monitoring suggest that recovery of electrophysiological brain activities anticipates, even by months, the recovery of consciousness and motor function. In general, however, the information provided by the conventional EEG parameters is limited [44–46].
A number of studies have specifically focused on the EEG reactivity, which reflects to a relevant extent the anatomic/functional integrity of cortex, reticular brainstem formation, diffuse thalamocortical projecting systems, and extrathalamic pathways from subthalamic and hypothalamic regions [47,48]. According to early and recent reports [49–51], EEG reactivity (possibly in association with evidence of stimulus-evoked sensory responses) is a reliable long-term predictor of outcome after brain injury, with a reported 92% predictive accuracy of EEG reactivity (98% in combination with Tc or MRI brain scans), compared to 72% of the Glasgow Coma Scale [51]. Computer-assisted methods for EEG quantitative analysis have been applied extensively in the investigation of EEG reactivity patterns and have documented effects of sensory stimulation otherwise undetectable after conventional visual EEG inspection [e. g. 52–54]. Analyses of the coherence between EEG signals recorded from distinct areas of the scalp (a measure of correlation in the frequencies domain) were applied to describe changes in the functional relationship among brain cortical structures (either interacting or driven by a common regulating structure) in patients with closed head injuries [55]. Davey et al. [56] measured interhemispheric coherence in a patient in persistent vegetative state after stroke, with destruction of right basal ganglia and thalamus and atrophic cerebral cortex of the same side documented by MRI. Damage in the left hemisphere was restricted to part of the posterior areas. Notable interhemispheric asymmetry was observed, suggesting that subcortical structures (primarily the basal ganglia and thalamus) regulate, at least in part, the coherence among cortical/subcortical structures involved in the generation of EEG signals. It has been proposed that changes in coherence induced by thalamocortical activation may sustain integrative forebrain function, with damage to these structures conceivably being at the basis of the pathophysiology of vegetative state.
EEG patterns peculiar to functional correlates of physiological sleep (such as spindle activity) are observed in patients in coma, regardless of depth, and are indicated as an index of favorable prognosis in spite of their still undefined pathophysiology [57]. In general, the appearance of a sleep — wake cycle (no matter how rudimentary) is correlated to a better outcome and is a common observation in the transition from coma to a vegetative state (Fig. 5.3) [58,59]. Evans and Bartlett [60] studied a population of 138 patients with brain injury, subdivided into classes according to a number of EEG and polygraphic parameters (wakefulness, sleeplike patterns, normal, abnormal, or absent arousal, etc.) and found the absence of any arousal activity to be highly predictive of development into a persistent vegetative state. D’Aleo et al. [61] observed that progressive restoration of the sleep — wake cycle and increase of rapid eye movement (REM) sleep are associated with clinical recovery from the vegetative state. Also, Bricolo and co-workers [62] report a correlation between REM sleep, non-REM sleep, sleep-like patterns, diffuse nonreactive high-amplitude delta (0.5–6.0 Hz) activity, etc., in repeating sleep recordings and the clinical course in 22 long-term comatose patients after a brain injury. These studies agree in indicating some correlation between the occurrence, degree of organization, and progressive organization of sleep patterns in longitudinal recordings and the development of the comatose state. There is evidence [63,64] of functional interaction between REM sleep and processes of memory consolidation (specifically of perception traces), which are affected by selective REM-sleep deprivation [65]. In perspective, increased REM sleep in patients recovering from the vegetative state [66] is a possible correlate of recovery of the memory mechanisms necessary for conscious cognitive functions.
Recently, researchers from the Loewenstein Rehabilitation Center in Tel Aviv [67] observed an association between sleep-related erection episodes and REM sleep (95% of episodes) in a group of patients who recovered from vegetative state. This association was comparable with that in healthy controls, with no differences in the duration and characteristics of both events. Although preliminary, this observation suggests that hypothalamic mechanisms may be unaffected in the vegetative state. Following a comparable approach, spontaneous nystagmus was observed by electro-oculographic recording methods in six patients in the vegetative state during wakefulness, phase I and REM sleep, but never in phase II and slow-wave sleep [68]. Although not predictive of outcome, the presence itself of the rapid phase of nystagmus implies some integrity of the paramedian pontine reticular formation and frontal cortex [69].
Stimulus-Related Evoked Potentials
Cortical and precortical brain structures respond to adequate sensory inputs also through changes in the electrophysiological activities (evoked potentials), which are time-locked to stimulus onset, depend on the dynamics of activation of stimulus-specific central nervous system (CNS) pathways/ structures, and reflect the mechanisms of sensory information processing. Visual, somatosensory and auditory stimulus-related potentials are used in clinical practice, as well as in animal and human research, following reliable methodological and interpretative standards [70].
Somatosensory Evoked Potentials
Somatosensory evoked potentials have been proposed as an acceptable predictor of output in early phases of coma [46,71,72], despite the differences existing between studies with regard, for example, to the study design, etiology of coma, age of patients, type of and time from brain injury (from coma to locked-in or apallic syndromes). The EEG and somatosensory evoked potentials were studied within 3 days of head injury in 100 patients [73] ranked according to their somatosensory responses (group 1, central conduction time within normal values bilaterally; group 2, central conduction time abnormally increased on one side; and group 3, cortical wave N20 undetectable on one side or both sides). The outcome was favorable in 90% of patients in group 1, 69% in group 2, and 6% in group 3, respectively. The EEG was not predictive, except in case of poor outcome for patients with isoelectric or severely abnormal EEG recordings. The prognostic values of EEG reactivity, somatosensory evoked potentials, and the Glasgow Coma Scale measured in the early days after coma onset were compared in 40 patients, 33 post-traumatic and seven post-anoxic [74]. The study reports a higher predictive value of the somatosensory evoked potentials (92%) compared to EEG reactivity (67%) and Glasgow Coma Scale (62%). The prediction of good outcome in post-traumatic patients with normal or borderline somatosensory evoked potentials, however, was not confirmed in post-anoxic patients, no matter how organized their somatosensory responses were. The latter observation suggests that etiology may play a role as a prognostic factor of outcome from coma and vegetative state, taking into account the several concomitant pathologies (such as brain edema or hydrocephalus) that may affect brain electrophysiology in the acute phases after brain injury. A study of the effects of brain injury and vegetative state on the somatosensory evoked potentials [75,76] did not detect any pattern common to all patients besides a normal response at Erb’s recording position excluding peripheral nerve damage, and a prolonged central conduction time, conceivably due to diffuse subcortical axonal injury, which was also documented at the postmortem examinations [77,78]. Interestingly enough, progressive clinical improvement and reduction of the central conduction time were concomitant in one patient, suggesting that recovery of sensory processes may be essential in order to regain a conscious state. Concomitant improvement of the clinical condition and somatosensory evoked potentials was also recently reported by Claassen and Hansen [79].
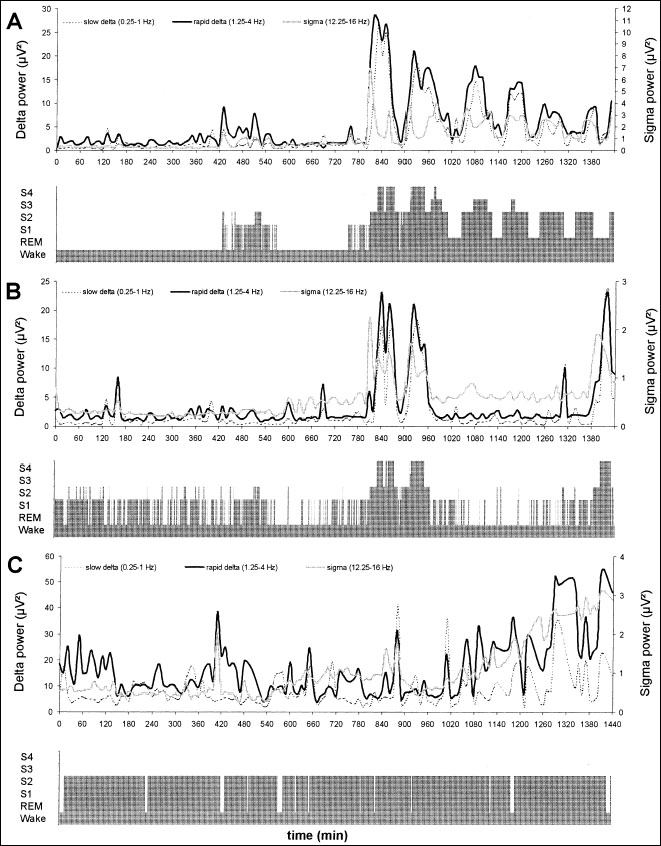
Fig 5.3 Hypnogram and time dynamics over a 24-h period of quantitative electroencephalography (EEG) in the 0.2–4.0 Hz (delta) and 12.2–16.0 Hz (sigma) activities of a normal individual (a) and two patients in permanent vegetative state (b, c). A short and rudimentary sleep organization is evident in b, with two cycles of the delta and sigma activities corresponding to episodes of deep sleep; the occurrence of consecutive deep sleep and delta activity cycles that are peculiar to the normal sleep-wake function (as seen in a) is truncated. No organized sleep—waking cycle can be recognized in c
Auditory Evoked Potentials
The brainstem evoked response to auditory stimulation is a compound potential generated within several structures of the brainstem auditory pathway [70]. In standard conditions, it is resistant to endogenous factors (e. g., ammonia, electrolytes, pH, PCO2) and exogenous factors (e. g., neuroactive drugs) [71,80,81], and it may therefore also be useful in metabolic coma. Changes in the latencies and conduction times between waves, high variability, changes in amplitude, or loss of individual waves can occur in different phases of acute midbrain syndromes. Recovery can be related to clinical improvement [82–84]. The reliability of brainstem auditory responses as a prognostic index is questioned, but a combined study of brainstem and middle-latency auditory responses appears to improve the prediction of outcome [71,85–87]. Frank et al. [88] reported on five nontraumatic comatose children, whose brainstem responses in the absence of a response to stimulation of the median nerve (reflecting thalamocortical network activation) was predictive of a permanent vegetative state.
Rappaport et al. [89] compared brainstem and long-latency auditory evoked responses in 75 patients with long-term traumatic brain injury. Increased latencies of long-latency responses proved to correlate significantly with the clinical outcome, while the brain-stem responses did not.
Jones et al. [90] measured the long-latency auditory responses to synthesized musical tones in 22 post-comatose patients. The presence of a response to complex tone stimuli was associated with discriminative hearing, i.e. the ability to respond differentially to verbal commands. The behavioral and electrophysiological findings were in agreement in 18 patients, suggesting that this approach may be suitable when identifying patients with “perception” capabilities in the presence of severe communication or motor disabilities.
Recently, Laureys et al. [91], in a PET study on patients in permanent vegetative state, documented a cascade of functional disconnection along the auditory cortical pathway, from the primary auditory area to parietal associative and limbic areas. The primary auditory cortex was activated by appropriate stimulation, and a brain-stem response was recorded.
Visual Evoked Potentials
Visual evoked potential studies in unresponsive patients mostly investigate the cortical/precortical responses to luminance stimuli, as investigation of the contrast visual function is not practicable in subjects who are unable to attend to the stimulation paradigm properly. Adler [92] studied these responses in 40 comatose patients in intensive care and assessed their relationship to intraventricular pressure. The complexity of the response (estimated as the number of detectable wave components) decreased with increasing depth of coma. Increased pressure resulted in a considerable reduction in the response amplitude, which recovered to baseline values after reduction of pressure. Clinical application of visual evoked responses in comatose patients is limited in comparison with somatosensory and auditory responses, and prediction of the clinical outcome is poor [71]. On the other hand, damage to the visual system, ranging from reduced visual acuity to visual field loss or even to functional blindness, often occurs after head injury [93], and visual evoked responses have proved useful in monitoring the recovery of visual function after trauma [94].
The definition of event-related potentials refers to a class of transient, phasic potentials elicited in conjunction with perceptual, cognitive, or motor events and regarded as concomitants of higher (‘cognitive”) brain functions. Unlike the responses to sensory stimulation, event-related potentials neither reflect the physical properties of stimulus nor depend on sensory input with physiological relevance, but result from situational links such as the significant association or omission, convergence, interaction, or unpredictable occurrence of significant events. In this respect, event-related potentials are, within limits, specific to the cognitive function explored, while sharing the sensitivity of electrophysiological measures. In the conventional experimental paradigms, conscious collaboration is required. However, studies in psychiatric or demented patients and animals have been performed successfully. The resulting evidence suggests that several physiological mechanisms concur in the generation of event-related potentials and reflect partly undefined neural functions that contribute to conscious behavior, but can occur independently [95–97]. Contingent negative variation (CNV) and P300 are classic examples of this, in a rapidly evolving field.
The CNV [98,99] is in agreement with theoretical models of operant conditioning. A negative steady potential shift with positive monotonic and inverted-U correlation with attention and arousal, respectively, the CNV is evoked by a temporal association between (conditional and imperative) stimuli. A motor response to the imperative stimulus is a rule. However, steady-state potentials comparable in shape to the CNV recorded in conscious subjects were obtained from two patients in deep coma (one of whom developed the vegetative state) by increasing the number of paired stimuli (Fig. 5.4) [100]. This observation is consistent with experimental evidence that steady shifts in membrane potential occur in the cortex and subcortical structures during memory processes, at levels of complexity of the involved neuronal assemblies that are not compatible with conscious operations [101–105].
A late positive response to attended stimuli, usually referred to as “P300” based on polarity and latency, is recorded at appropriate scalp locations when stimuli in the same sensory modality, but with differing physical characteristics and frequency of occurrence, are presented and the subject is requested to attend the infrequent stimulus. The generators are unknown, but experimental evidence suggests interaction of cortical/subcortical structures such as the limbic system, thalamus, corpus callosum, and distributed cortical areas [106–113]. Tomberg and Desmedt [114] suggest that the P300 may reflect time-related inhibition (possibly due to subcortical activation) of the neuronal functional assemblies involved in the processing of attended stimuli. Kotchoubey et al. [115] reported observing P300 responses in 14 patients in the vegetative state and 19 patients who had recovered to the condition of minimal response. In all cases, conventional brain-stem acoustic responses made it possible to exclude significant impairment of the precortical auditory system. Three different acoustic stimulation paradigms were applied: two (target and nontarget) sinusoidal tones, two complex tones, and two vowels (an “O” in a female voice and an “I” in a male voice). In both groups of patients, the P300 response was observed more frequently when the complex paradigms were applied. Comparable results were obtained in patients in permanent vegetative state when verbal paradigms with affective-emotional connotations, eliciting a P300 (e. g., the patient’s names compared to other names) were compared with the classic oddball paradigms [116]. Keren and co-workers [117] reported on 16 severely brain-injured patients, whose P300 and neuropsychological assessments were performed 1, 2, and 5 months after the trauma. At the beginning of follow-up, the P300 latencies were significantly longer in patients with lower Glasgow Coma Scale scores. A progressive reduction in the P300 latencies was observed in patients with low scores, with the differences from patients with high scores disappearing at a later time in parallel with improvements in cognitive performance as detected by neuropsychological tests. These observations suggest a possible clinical use of event-related potentials as an index of brain activity relating to the recovery from coma or vegetative state, as well as some participation of limbic structures (involved in the “emotional recognition” of human voice and names) in the generation of a P300 response in vegetative patients and, conceivably, in healthy subjects. However, a prediction of favorable outcome based on the observation of a P300 response is still speculative.
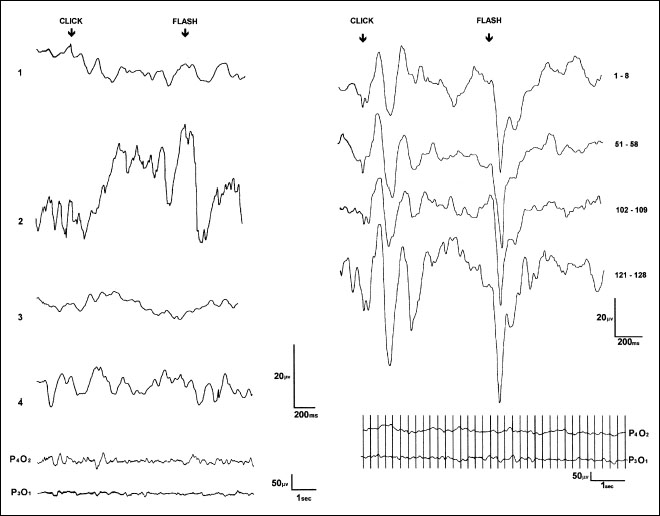
Fig 5.4 A negative steady potential shift resembling contingent negative variation (CNV), recorded by conventional methods in two patients in deep coma (see the original report for details). On the left are shown: 1) the absence of response to a first standard sequence of eight paired stimuli (sufficient to evoke CNV in healthy subjects) and the averaged response to eight paired stimuli obtained after a long series of stimuli (about 150) as recorded from the vertex (2) and mid-parietal areas (3). On the right are shown the averaged responses to sequences of eight paired stimuli, with a CNV-like response appearing only after a long series of stimuli. (Reproduced from Electroencephalography and Clinical Neurophysiology [100], with permission from Elsevier Science)
Jennett and Plum [45] introduced the term “persistent vegetative state” to describe the condition of “wakefulness without awareness” following brain injuries. The cortical-subcortical structures involved in the regulation of vigilance also modulate cortical electrophysiology. An electrophysiological approach to the understanding of the mechanisms of coma and vegetative state therefore appeared conceptually and methodologically appropriate and was favored. However, research in this field did not expand and become systematic, as in other areas of clinical neurology (e.g., epilepsy, dementia, movement or vascular disorders, etc.), and the available information is often derived from case reports or studies in small groups of patients. Methodologies have often differed among studies, making meta-analyses impracticable, and prospective study designs are exceptional.
The approach nevertheless still appears promising, and it seems realistic to expect that more refined and controlled methodologies and research strategies may help predict the outcome and allow physicians to allocate resources. Remarkably, questions about the physiological concomitants of consciousness have expanded beyond the fields of philosophy and epistemology and are becoming key issues in today’s neuroscience, as well as in neuroinformatics and in research on artificial intelligence and robotics. Improved and systematic neurophysiological investigations combined with up-to-date functional neuroimaging techniques — such as functional MRI (fMRI) and PET — as well as collaboration between neuroscientists and anesthesiologists, should be regarded as a sound approach to understanding CNS function, the mechanisms regulating vigilance and allowing conscious behavior, and the pathophysiology of the conditions of impaired consciousness that can follow brain injuries.
References
1. De Salles AAF, Muizelaar JP, Young HF. Hyperglycemia, cerebrospinal fluid lactic acidosis, and cerebral blood flow in severely head-injured patients. Neurosurgery 1987; 21: 45–50.
2. Zupping R, Magi M, Tikk A et al. Cerebral metabolic disorders during prolonged unconsciousness after severe head injury. Eur Neurol 1972; 9: 145–50.
3. Sazbon L, Solzi P, Berghaus N. Periarticular new bone formation in hemiplegic patients. Harefuah 1980; 99: 317–8.
4. Becker E, Sazbon L, Najenson T. Gastrointestinal haemorrhage in long lasting traumatic coma. Scand J Rehabil Med 1978; 10: 23–6.
5. Hamil RW, Woolf PD, McDonald JV, et al. Catecholamines predict outcome in traumatic brain injury. Ann Neurol 1987; 21: 438–43.
6. Hans P, Born JD, Albert A. Extrapolated creatine kinase-BB isoenzyme activity in assessment of initial brain damage after severe head injury. J Neurosurg 1987; 66: 714–7.
7. Rabow L, Dec Salles AAF, Becker DP, et al. CSF brain creatinine kinase levels and lactic acidosis in severe head injury. J Neurosurg 1986; 65: 625–9.
8. Bakay RAE, Ward AA. Enzymatic changes in serum and cerebrospinal fluid in neurological injury. J Neurosurg 1983; 58: 27–37.
9. Rudman D, Fleischer AS, Kutner MH, Raggio JF. Suprahypophyseal hypogonadism and hypothyroidism during prolonged coma after head injury. J Clin Endocrinol Metab 1977; 45: 747–54.
10. Sazbon L. Prolonged coma. Prog Clin Neurosci 1985; 2: 65–81.
11. Najenson T, Sazbon L, Fiselson J, et al. Recovery of communicative functions after prolonged traumatic coma. Scand J Rehabil Med 1978; 10: 15–21.
12. Woolf, PD, Hamill RW, Lee RA, et al. The predictive value of catecholamines in assessing outcome in traumatic brain injury. J Neurosurg 1987; 66: 875–82.
13. Woolf PD, Cox C, Kelly M, et al. The adrenocortical response to brain injury. Alcohol Clin Exp Res 1990; 14: 917–21.
14. Pezzili R, Billi P, Barakat B, et al. Serum pancreatic enzymes in patients with coma due to head injury or acute stroke. Int J Clin Lab Res 1997; 27: 244–6.
15. Sazbon L. [Personal communication].
16. Kapaki E, Segditsa J, Papageorgiou C. Zinc, copper and magnesium concentration in serum and CSF of patients with neurological disorders. Acta Neurol Scand 1989; 79: 373–8.
17. McClain CJ, Twyman DL, Ott LG, et al. Serum and urine zinc response in head- injured patients. J Neurosurg 1986; 64: 224–30.
18. Sorbi S, Nacmias B, Piacentini S, et al. ApoE as a prognostic factor for post-traumatic coma [letter]. Nature Med 1995; 1: 852.
19. Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E e4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology 1999; 52: 244–8.
20. Roses AD, Saunders A. Head injury, amyloid and Alzheimer disease. Nature Med 1995; 1: 603–4.
21. Adams JH. Brain damage in fatal nonmissile head injury in man. In: Vinken PJ, Bruyn GW, editors. Handbook of clinical neurology, vol 53. Amsterdam: North-Holland, 1990: 43.
22. Kinney HC, Samuels MA. Neuropathology of the persistent vegetative state: a review. J Neuropathol Exp Neurol 1994; 53: 548–58.
23. Zimmermann RA, Bilaniuk LT, Genneralli T. Computed tomography of shearing injuries of the cerebral white matter. Radiology 1978; 127: 393–6.
24. Vallasciani M, Gironelli L, Tulli D, Chiaramoni L, Salvolini U. Neuroradiological investigations in patients with persistent vegetative state. Rev Neuroradiol 2000; 13 (Suppl 2 ): 81.
25. Santoro G, Lanza PL. Traumi cranio-encefalici. [Personal communication].
26. Hijdra A, Zandbergen EG, de Haan RJ. PET scanning and neuronal loss in acute vegetative state. Lancet 2000; 355: 1826.
27. Reider-Groswasser I, Costeff H, Sazbon L, Groswasser Z. CT findings in persistent vegetative state following blunt traumatic brain injury. Brain Inj 1997;11: 865–70.
28. Kampfl A, Schmutzhard E, Franz G, et al. Prediction of recovery from post-traumatic vegetative state with cerebral magnetic resonance imaging. Lancet 1998;351: 1763–7.
29. Kampfl A, Franz G, Aichner F, et al. The persistent vegetative state after closed head injury: clinical and magnetic resonance imaging findings in 42 patients. J Neurosurg 1998; 88: 809–16.
30. Gieron MA, Korthals JK, Riggs CD. Diffuse axonal injury without direct head trauma and with delayed onset of coma. Pediatr Neurol 1998; 19: 382–4.
31. Ricci R, Barbarella G, Musi P, Boldrini P, Treviasn C, Basaglia N. Localized proton MR spectroscopy of brain metabolism changes in vegetative patients. Neuroradiology 1997; 39: 313–9.
32. Sinson G, Bagley LJ, Cecil KM, et al. Magnetization transfer imaging and proton MR spectroscopy in the evaluation of axonal injury: correlation with clinical outcome after traumatic brain injury. Am J Neuroradiol 2001; 22: 143–51.
34. Petsche H, Pockberger H, Rappelsberger P. On the search for the sources of the electroencephalogram. Neuroscience 1984; 11: 1–27.
35. Elul R. The genesis of the EEG. Int Rev Neurobiol 1971; 15: 227–72.
36. Speckmann EJ, Caspers H, editors. Origin of cerebral field potentials. Stuttgart: Thieme, 1979.
37. Pilgreen KL. Physiologic, medical and cognitive correlates of electroencephalography. In: Nunez P, editor. Neocortical dynamics and human EEG rhythms. New York: Oxford University Press, 1995: 195–248.
38. Steriade M. Corticothalamic resonance, states of vigilance and mentation Neuroscience 2000; 101: 243–76.
39. Niedermayer E, editor. Electroencephalography: basic principles, clinical applications and related fields. Baltimore: Williams and Wilkins, 1993.
40. Coenen AM. Neuronal phenomena associated with vigilance and consciousness: from cellular mechanisms to electroencephalographic patterns. Conscious Cogn 1998; 7: 42–53.
41. Celesia GG. EEG and coma: is there a prognostic role for EEG? Clin Neurophysiol 1999; 110: 203–4.
42. Plum F, Posner JB, editors. The diagnosis of stupor and coma. Philadelphia: FADC, 1980.
43. Dolce G, Kaemmerer E. Contributo anatomo clinico alla conoscenza della sindrome apallica. Sistema Nervoso 1967; 19: 12–23.
44. Lucking CH, Mullner E, Pateisky K, Gerstend-brand F. Electroencephalographic findings in apallic syndrome. In: Dalle Ore G, Gerstend-brand F, Lucking CH, Peters G, Peters UH, editors. The apallic syndrome. New York: Springer, 1977: 144–54.
45. Jennett B, Plum F. Persistent vegetative state after brain damage. Lancet 1972; i: 734–7.
46. Chatrian G, Bergamasco B, Bricolo A, Frost JD, Prior P. IFCN recommended standards for elctrophysiologic monitoring in comatose and other unresponsive states: report of an IFCN committee. Electroencephalogr Clin Neurophysiol 1996; 99: 103–23.
47. Steriade M, Buzsàki G. Parallel activation of thalamic and cortical neurons by brainstem and basal forebrain cholinergic systems. In: Steriade M, Biesold S, editors. Brain cholinergic systems. Oxford: Oxford University Press, 1990: 3.
48. Steriade M, Jones EG, Llinas RR, editors. Thalamic oscillations and signaling. New York : Wiley, 1990.
49. Chatrian G. Electroencephalographic patterns resembling those of sleep in certain comatose states after injuries to the head. Electroencephalogr Clin Neurophysiol 1963; 15: 272–80.
50. Courjon J, Naquet R. Value of the EEG in diagnosis and prognosis immediately after cranial trauma. Rev EEG Neurophysiol 1971; 1: 133–50.
51. Gutling E, Gosner A, Imhof HG, Landis T. EEG reactivity in the prognosis of severe head injury. Neurology 1995; 45: 915–8.
52. Ferrillo F, Rivano C, Rosadini G, Rossi GF, Turella C. EEG spectral analysis in coma. Electroencephalogr Clin Neurophysiol 1969; 27: 700.
53. Rosadini G, Ferrillo F. Automatic analysis of spontaneous EEG activity in the functional exploration of the human brain. In: Somjen GG, editor. Neurophysiology studied in man: proceedings of a symposium held in Paris at the Faculté des Sciences, 20–22 July 1971. Amsterdam: Excerpta Medica, 1972: 453–460.
54. Sannita WG, Rosadini G. Computerized EEG analysis in the management of comatose patients: an operational proposal. In: Lechner H, Aranibar A, editors. EEG and clinical neurophysiology. Amsterdam: Excepta Medica, 1980: 583–91.
55. Tatcher RW, Cantor DS, McAlaster R, Geisler F, Krause P. Comprehensive predictions of outcome in closed head injured patients: the development of prognostic equations. Ann NY Acad Sci 1991; 620: 82–101.
56. Davey M, Victor J, Schiff N. Power spectra and coherence in the EEG of a vegetative patient with severe asymmetric brain damage. Clin Neurophysiol 2000;111: 1949–54.
57. Kaplan PW, Genoud D, Ho TW, Jallon P. Clinical correlates and prognosis in early spindle coma. Clin Neurophysiol 2000; 111: 584–90.
58. Gerstenbrand F. Das traumatische apalliche syndrom. Vienna: Springer, 1967: 344.
59. Zeman A. Persistent vegetative state. Lancet 1997; 350: 795–9.
60. Evans BM, Bartlett JR. Prediction of outcome in severe head injury based on recognition of sleep related activity in the polygraphic electroencephalogram. J Neurol Neuros Psych 1995; 59: 17–25.
61. D’Aleo G, Saltuari L, Gerstenbrand F, Bramanti P. Sleep in the last remission stages of vegetative state of traumatic nature. Funct Neurol 1994; 9: 189–92.
62. Bricolo A, Gentilomo A, Rosadini G, Rossi F. Long lasting post-traumatic unconsciousness. Acta Neurol Scand 1968; 44: 512–32.
63. Crick F, Mitchinson G. The function of dream sleep. Nature 1983; 304: 111–4.
64. Kavanau JL. Memory, sleep and the evolution of mechanisms of synaptic efficacy maintenance. Neuroscience 1997; 79: 7–44.
65. Karni A, Tanne D, Rubenstain BS, Askenasy JJ, Sagi D. Dependence on REM sleep of overnight improvement of a perceptual skill. Science 1994; 265: 679–82.
66. D’Aleo G, Bramanti P, Silvestri R, Saltuari L, Gerstenbrand F, Di Perri R. Sleep spindles in the initial stages of the vegetative state. Ital J Neurol Sci 1994; 15: 347–51.
67. Oksenberg A, Arons E, Sazbon L, Mizrahi A, Radwan H. Sleep-related erections in vegetative state patients. Sleep 2000; 23: 953–7.
68. Gordon C, Oksenberg A. Spontaneous nystagmus across the sleep-wake cycle in vegetative state patients. Electroencephalogr Clin Neurophysiol 1993; 86: 132–7.
69. Leigh R, Zee D. The neurology of eye movements. Philadelphia: Davis, 1983.
70. Regan D. Human brain electrophysiology: evoked potentials and evoked magnetic fields in science and medicine. New York: Elsevier, 1989.
71. Greenberg RP, Mayer DJ, Becker DP, Miller JD. Evaluation of brain function in severe human head trauma with multimodality evoked potentials. J Neurosurg 1977; 47: 150–77.
72. Sleigh JW, Havill JH, Frith R, Kersel D, Marsh N, Ulyatt D. Somatosensory evoked potentials in severe traumatic brain injury: a blinded study. J Neurosurg 1999; 91: 577–80.
73. Hutchinson DO, Frith RW, Shaw NA, Judson JA, Cant BR. A comparison between electroencephalography and somatosensory evoked potentials for outcome prediction following severe head injury. Electroencephalogr Clin Neurophysiol 1991; 78: 228–33.
74. Piersanti P, Cesaretti C, Matà S, et al. Reattività EEG e potenziali evocati somatosensitivi. Confronto e possibili discordanze nella prognosi precoce del coma grave. In: Rossi B, Gabrielli L, Murri L, editors. Atti del VI Congresso della Società Italiana di Psicofisiologia, Pisa, Italy, 1996: 49–57.
75. Keren O, Groswasser Z, Sazbon L, Ring C. Somatosensory evoked potentials in prolonged post-comatose unawareness state following traumatic brain injury. Brain Inj 1991; 3: 233–40.
76. Keren O, Sazbon L, Groswasser Z, Shmuel M. Follow-up studies of somatosensory evoked potentials and auditory evoked potentials in patients with post-coma unawareness of traumatic brain injury. Brain Inj 1994; 8: 239–47.
77. Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to non-missile head injury in humans: analysis of 45 cases. Ann Neurol 1982; 27: 557–63.
78. Adams JH, Graham DI, Gennarelli TA, Maxwell WL. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry 1991; 54: 481–3.
79. Claassen J, Hansen HC. Early recovery after closed traumatic head injury: somatosensory evoked potentials and clinical findings. Crit Care Med 2001; 29: 494–502.
80. Sutton LN, Frewen T, Mausch R, Jaggi J, Bruce DA. The effect of deep barbiturate coma on multimodality evoked potentials. J Neurosurg 1982; 57: 178–85
81. Sannita WG. Stimulus-related evoked potentials: from basic science to clinical neuropharmacology. Electroencephalogr Clin Neurophysiol Suppl 1996; 46: 95–106.
82. Rumpl E, Prugger M, Gerstenbrand F, Brunnhuber W, Badry F, Hackl JM. Central somatosensory conduction time and acoustic brainstem transmission time in post-traumatic coma. J Clin Neurophysiol 1988; 3: 237–60.
83. Schwarz G, Litscher G, Rumpl E, Pfurtscheller G, Reimann R. Brainstem auditory evoked potentials in respiratory insufficiency following encephalitis. Int J Neurosci 1996; 84: 35–44.
84. Werner RA, Vanderzant CW. Multimodality evoked potentials testing in acute mild closed head injury. Arch Phys Med Rehabil 1991; 72: 31–4.
85. Litscher G. Middle latency auditory evoked potentials in intensive care patients and normal controls. Int J Neurosci 1995; 83: 253–67.
86. Sazabon L, Solzi P, Steinvil Y, Becker E. Blink reflex in patients in prolonged comatose state. Electromyogr Clin Neurophysiol 1988; 28: 151–8.
87. Hall JW, Mackey-Hargadine J, Allen SJ. Monitoring neurological status of comatose patients in the intensive care unit. In: Jacobson JT, editor. The auditory brainstem response. San Diego: College Hill Press, 1985: 253–83.
88. Frank M, Furgiuele T, Etheridge J. Predicting of chronic vegetative state in children using evoked potentials. Neurology 1985; 35: 931–4.
89. Rappaport M, Hemmerle AV, Rappaport ML. Short and long latency auditory evoked potentials in traumatic brain injury patients. Clin Electroencephalogr 1991; 22: 199–202.
90. Jones SJ, Vaz Pato M, Sprague L, Stokes M, Munday R, Haque N. Auditory evoked potentials to spectro-temporal modulation of complex tones in normal subjects and patients with severe brain injury. Brain 2000; 123: 1007–16.
91. Laureys S, Faymonville ME, Degueldre C, et al. Auditory processing in the vegetative state. Brain 2000; 123: 1589–601.
92. Adler G, Bransi A, Prange HW. Neuro-monitoring using visual evoked potentials in comatose neurologic intensive care patients. EEG EMG Z Elektroenzephalogr Elektromyogr Verwandte Geb 1991; 22: 254–8.
93. Tierney DW. Visual dysfunction in closed head injury. J Am Optom Assoc 1988; 59: 614–22.
94. Freed S, Hellerstein LF. Visual electrodiagnostic findings in mild traumatic brain injury. Brain Inj 1997; 11: 25–36.
95. Johnson R, Rohrbaugh JW, Parasuraman R, editors. Currents trend in event-related potentials research. Electroencephalogr Clin Neurophysiol Suppl 1987; 40 [special issue].
96. Brunia CHM, Mulder G, Verbaten MN, editors. Event related brain research. Electroencephalogr Clin Neurophysiol Suppl 1991; 42 [special issue].
97. Naatanen R. Attention and brain function. Hillsdale, NJ: Erlbaum, 1992.
98. Walter WG, Cooper R, Aldridge VJ, McCallum WC, Winter AL. Contingency negative variation: an electric sign of sensori-motor association and expectancy in the human brain. Nature 1964; 203: 380–4.
99. Tecce JJ, Savignano-Bowman J, Cole JO. Drug effects on CNV and eyeblinks: the distraction arousal hypothesis. In: Lipton MA, DiMascio A, Killam KF, editors. Psychopharmacology: a generation of progress. New York: Raven Press, 1978: 745–58.
100. Dolce G, Sannita W. A CNV-like negative shift in deep coma. Electroencephalogr Clin Neurophysiol 1973; 34: 647–50.
101. John ER, Herrington RN, Sutton S. Effects of visual form on the evoked response. Science 1967; 155: 1439–42.
102. Horel JA, Vierck CJ Jr, Pribram KH, Spinelli DN, John ER, Ruchkin DS. Average evoked responses and learning. Science 1967; 158: 394–5.
103. Albright TD, Kandel ER, Posner MI. Cognitive neuroscience. Curr Opin Neurobiol 2000; 10: 612–24.
104. Spencer WA, Kandel ER. Cellular and integrative properties of the hippocampal pyramidal cell and the comparative electrophysiology of cortical neurons. Int J Neurol 1968; 6: 266–96.
105. Kandel ER, Hawkins RD. The biological basis of learning and individuality. Sci Am 1992; 267: 78–86.
106. Polich J, Squire LR. P300 from amnesic patients with bilateral hippocampal lesions. Electroencephalogr Clin Neurophysiol 1993; 86: 408–17.
107. Ji J, Porjesz B, Begleiter H, Chorlian D. P300: the similarities and differences in the scalp distribution of visual and auditory modality. Brain Topogr 1999; 11: 315–27.
108. Katayama J, Polich J. Auditory and visual P300 topography from a 3 stimulus paradigm. Clin Neurophysiol 1999; 110: 463–8.
109. Frodl-Bauch T, Bottlender R, Hegerl U. Neurochemical substrates and neuroanatomical generators of the event-related P300. Neuropsychobiology 1999; 40: 86–94.
110. Buchner H, Gobbele R, Waberski TD, Wagner M, Fuchs M. Evidence for independent thalamic and cortical sources involved in the generation of the visual 40 Hz response in humans. Neurosci Lett 1999; 269: 59–62.
111. Clark VP, Fannon S, Lai S, Benson R, Bauer L. Responses to rare visual target and distractor stimuli using event-related fMRI. J Neurophysiol 2000; 83: 3133–9.
112. Trinka E, Pfisterer G, Unterrainer J, et al. Multimodal event-related potential P3 after transient global amnesia. Eur J Neurol 2000; 7: 81–5.
113. Trinka E, Unterrainer J, Staffen W, Loscher NW, Ladurner G. Delayed visual P3 in unilateral thalamic stroke. Eur J Neurol 2000; 7: 517–22.
114. Tomberg C, Desmedt JE. Human perceptual processing: inhibition of transient prefrontalparietal 40 Hz binding at P300 onset documented in non-averaged cognitive brain potentials. Neurosci Lett 1998; 255: 163–6.
115. Kotchoubey B, Lang S, Baales R, et al. Brain potentials in human patients with extremely severe diffuse brain damage. Neurosci Lett 2001; 301: 27–40.
116. Giorgianni R, Lo Presti R, D’Aleo G, Mondo N, Di Bella P, Bramanti P. Event-related potentials in patients in persistent vegetative state. Electroencephalogr Clin Neurophysiol 1997; 103: 149.
117. Keren O, Ben Dror S, Stern MJ, Goldberg G, Grosswasser Z. Event related potentials as an index of cognitive function during recovery from severe closed head injury. J Head Trauma Rehabil 1998; 13: 15–30.
< div class='tao-gold-member'>




