2 Preliminary Concepts
Leon Sazbon and Giuliano Dolce
Etiology
Vegetative state (VS) is defined behaviorally as the absence of an adequate response to the outside world and absence of any evidence of reception or projection of information in the presence of sleep – wake cycles. Patients may have periods of wake-fulness with open eyes and movement, but responsiveness is limited to primitive postural and reflex movements of the limbs. They never speak, and some never regain recognizable mental function [1]. According to the Oxford English Dictionary [2], individuals in VS “live a merely physical life, devoid of intellectual activity or social intercourse; [they are] capable of growth and development but devoid of sensation and thought.” The term was coined in 1972 by Jennett and Plum [1] to eliminate the confusion caused by the multitude of disorders in the literature, such as akinetic mutism, apallic syndrome, and prolonged coma, which share the common denominators of wakefulness with unawareness. These clinical criteria for VS were more recently confirmed by the Multi-Society Task Force [3].
Over the years, certain qualifiers have been added to the term VS, often incorrectly. The term “persistent vegetative state” (PVS) has been used by some researchers to refer to any type of VS, and by others to refer specifically to VS of long duration [3–6]. The word “persistent” is defined by Webster’s Tenth Collegiate Dictionary [7] as “continuing to exist in spite of interference or treatment”, and “persistence” is defined by the COBUILD Collins English Language Dictionary [8] as continuing “to exist, even after you have tried to make it disappear.” The indiscriminate use of “persistent” in relation to VS has led to the misconception that the syndrome is always irreversible, when the term actually refers merely to a disorder that began in the past and is continuing in the present. This problem is exacerbated when (as is not uncommon), “persistent” is replaced with “permanent”. The latter term describes to a state that began in the present and will continue through the future – implying a negative and irreversible prognosis. There is a fine line between reversible and the irreversible VS, and a clear clinical definition is still lacking. On the basis of our experience, we consider VS, using practical criteria, irreversible when it has persisted for 1 year in patients after coma of traumatic origin, or for 6 months in patients after coma of non-traumatic etiology. These distinctions have importance for medical decisions regarding the introduction or continuation of therapy and its intensity. The International Working Party [9] has recommended that both terms – persistent and permanent – should be dropped altogether.
VS should be differentiated from coma, which is characterized by an inability to obey commands, utter recognizable words, or open the eyes, and the absence of sleep – wake cycles. Coma is always a transient state. According to Plum [10], comatose patients who survive for more than a few days or weeks will regain cyclic electroencephalographic patterns of arousal/nonarousal with or without their behavioral appearance. These patients may live indefinitely, provided they retain hypothalamic functions and a majority of tegmental brain-stem functions [10].
VS is always an expression of a direct primary brain pathology and is not an extension of coma. Although it exists from the start, the VS is masked by the state of coma, thus hindering diagnosis.
The causes of VS may be acute (traumatic or non-traumatic) or chronic (degenerative and metabolic disorders or developmental malformations). Examples of traumatic injury are motor vehicle accidents, gunshot wounds, domestic accidents, and birth injury; nontraumatic causes include hypoxic – ischemic encephalopathy, central nervous system (CNS) infection, CNS tumor, cerebrovascular injury, and CNS toxins or poisoning. Examples of degenerative disorders are Alzheimer’s disease, multi-infarct dementia, and Creutzfeldt-Jakob disease, and examples of developmental malformations include congenital hydrocephalus and severe microcephaly. The most common acute causes in adults and children are injury-induced head trauma and hypoxic-ischemic encephalopathy, generally following coma of several days to weeks. In rare cases, VS may occur immediately after traumatic injury (documented, for example, in boxers after a knockout). The duration of VS can vary widely, from only a few seconds to many years.
Epidemiology
The early editions of the International Classification of Diseases (ICD) grouped vegetative state (VS) with acute coma (code 780.01), and it was only 9 years ago that VS was first listed as a distinct entity (code 780.03). No epidemiological studies of VS have been conducted since the new classification, so figures are hard to establish. The estimated annual incidence of brain trauma in the United States varies widely, from 180 to 400 patients per 100 000 population [11–13]. The prevalence of VS is unknown, although in some American publications the suspicion is stated that the numbers range between 10 000 and 25 000 adults and 6000 to 10 000 children [14–18]. Researchers in Japan, France, Italy and other countries have calculated a national annual incidence of 0.9–2 per 100 000 [19–22]. The percentage of patients remaining in VS has been estimated in different studies to range from 0.2 % to 14 % of all cases of acute coma in various studies [19,23–28], depending on patient age, etiology, and the temporal criteria used. The actual rate may be even higher, as patients with open eyes are often misdiagnosed as being comatose.
In Israel, until recently, all patients in the country who were in traumatic VS for at least 1 month were referred to the Loewenstein Rehabilitation Hospital. This center therefore provides an excellent setting for an epidemiological analysis of VS in Israel. From 1975 to 1998, a total of 580 patients with VS of traumatic origin were admitted, an average of 23.2 new cases per year. The true incidence may of course be higher, as these figures exclude patients with short-term VS. For the first 8 years (1975–1983), an average of 16.3 patients were admitted per year, whereas in the last 15 years, this number jumped to 30. This increase may be explained by the increased use of intensive life support already at the accident site, the more rapid means of transport to neurosurgical centers, and the accelerated growth of the population. According to Israeli population studies, the incidence of traumatic VS is in the order of 0.4–0.5 per 100 000 inhabitants [29,30]
Age and Gender
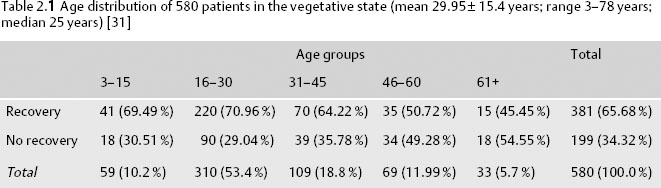
The age distribution of patients in VS was calculated in an Israeli series of 580 patients at the Loewenstein Hospital and correlated with recovery [31]. The findings are shown in Table 2.1.
There were 446 males and 134 females, a ratio of 3.3 : 1.0. In an earlier series of 134 patients reported by the same authors [30], the mean age was 26.8 ± 14.6 years, with a range of 3–79 and a male/female ratio of 4 : 1. Most of the cases of VS (72.24 %) occurred in patients between the ages of 16 and 45.
Age was apparently related to recovery of consciousness, with the highest recovery rate (about 70 %) being recorded in patients less than 30 years old, followed closely by the 31–45-year-old group. Only 50.5 % of patients aged over 46 years recovered consciousness. The mean age in the recovered group was 28.1 ± 14.2 years, and in the nonrecovered group, 33.4 ± 16.9 years; this difference was statistically significant (P > 0.001) [31]. However, in our experience, children have a poorer functional outcome than adults. Some authors hypothesize that the lesser myelination in the young brain and its lower ability to regulate vascular perfusion make it subject to greater shearing forces during acute injury [32–34]. However, other authors disagree, claiming that the chances for both vital and functional recovery decrease with age [35,36].
The rate of occurrence of new cases of VS shows seasonal variability. The highest frequency has been noted in fall (autumn), followed in order by summer, winter, and spring [30,37]. In the Israeli study by Sazbon et al. [20], the frequency peaked in October and August (P < 0.003), coinciding with the school holiday and the Jewish High Holy Days. Time of day is also a factor, with 70 % of patients being injured between 5 p. m. and midnight [30].
Circumstances of VS
Most cases of VS can be traced to traumatic head injury. In Israel, the most common cause of traumatic head injury leading to VS is road accidents (Table 2.2), with a considerably higher rate of blunt injuries (95 %) than penetrating injuries (5 %).
Sazbon et al. [30] studied 70 patients in VS admitted to Loewenstein Rehabilitation Hospital who were victims of car accidents. They found that 39 had been pedestrians, 36% passengers in the vehicle, 16% drivers of the vehicle, and 9% cyclists. Interestingly, the pedestrians received only 17–21 % of the acute brain injuries and the passengers 19–21 %, whereas the drivers received 37 % of the acute head injuries, but accounted for only 16% of the VS cases. The pedestrians and cyclists had the worst prognosis, with 56% and 50%, respectively, remaining in permanent VS until death. Rates for drivers and passengers were 40 % and 37 %, respectively. These data are in line with the findings of the 1988 Fédération Française des Associations de Médecins Conseils (FFAMC) conference [38], organized by consultant physicians of insurance companies in France, which concluded that VS lasted markedly longer in pedestrians hit by cars than in car drivers.
| 1975–1983 | 1975–1998 | |
| (n = 134) | (n = 580) | |
| Road accidents | 92(69%) | 480(79.32%) |
| Warwounds and army accidents | 16(12%) | 46(7.93%) |
| Miscellaneous* 2 | 26(19%) | 74(12.75%) |
* Assaults, falls, domestic mishaps, other accidents. This rate may be lower than in other countries be-cause of the low incidence of alcoholism in Israel [30]. Reproduced with permission from Taylor & Francis Limited, http//www.tandf.co.uK. Brain Injury 6: 359–362,1992
Recovery
Recovery from VS is defined as the ability to establish visual or verbal contact with the outside world [32]. Sazbon and Grosswasser [29] reported a 54% recovery rate in their earlier series of 134 patients and 65.68% in the later series of 580 patients [31]. Most patients who regained consciousness did so in the first 3 months. After 1 year, only a handful of the patients still in VS achieved a state of minimal responsiveness. The others remained in permanent VS or died.
Regrettably, no large series of VS patients have recently been published in the medical literature for comparison. Only two studies of VS of traumatic etiology were conducted in the 1970s – those by Vigouroux et al. [39] with 150 cases and Higashi et al. [19] with 100 cases of all etiologies, of which 38% were of traumatic origin. In the 1980s, studies were conducted by Bricolo et al. [23] (135 cases), Brule et al. [20] (100 cases), Lyle et al. [40] (159 cases), and Braakman and Jennett [27] (140 cases). In the 1990s, Sazbon and Grosswasser [29] published one series of 134 cases of traumatic etiology, followed by Levin et al. [41] (93 cases) and the Multi-Society Task Force on PVS publication, which summarized previous publications [17]. To date, there have been only three studies of patients in VS of nontraumatic origin: Levy et al. [42] (55 cases), Heind and Laub [43] (127 cases of mixed etiology) and Sazbon et al. [44] (102 cases).
Neuropathological Basis of the Vegetative State
Research into the neuropathology of vegetative state (VS) has been delayed because of a low level of interest among physicians and the few centers working with this type of patient worldwide.
Patients with VS appear to have a macroscopically normal brain. In 1956, Strich was the first to suggest that diffuse axonal injury (DAI) in the sub-cortical white matter and descending tracts into the brain stem was the basis of what he termed “severe post-traumatic dementia” [45]. This was later supported by Peerless and Rewcastle [46] and Zimmerman et al. [47]. However, Jellinger [48] and Peters and Rothemund [49] claimed that the axonal damage is secondary ischemia, brain swelling, or high intracranial pressure. In animal studies conducted in the 1980s, Gennarelli et al. [50] and Adams et al. [51] proved that the primary damage was axonal.
Most of our current knowledge on the neuropathology of VS comes from the studies by Adams et al. [52–54], Graham et al. [55], and Kinney and colleagues [56,57]. Recently, Adams et al. [58] conducted a precise and detailed neuropathological brain study of 49 patients in VS who never recovered, including 35 after blunt head injury. They found, in agreement with Kinney and Samuels [56], that VS results from widespread and bilateral damage to the cerebral cortex itself, the thalamus, and many of the intracortical and subcortical connections. This leads to one of three presentations: diffuse destruction of the cerebral cortex, either alone or associated with diffuse damage to the white matter; or diffuse damage to the thalamus only. By contrast, Maxwell et al. [59] claimed that the diffuse axonal damage was in effect a manifestation of a process of secondary axotomy, in which acute brain injury caused focal swelling of the axons and mitochondria, nodal blebs, and a focal decrease in the internodal axonal diameter, followed by loss of axonal microtubules, alterations in neurofilaments, involution of the internodal axolemma, and development of axonal swelling, culminating in axonal separation with axonal retraction bulbs. There may be microcystic spaces, with residual hemosiderin and macrophages [60]. The primary axotomy is identified within 1 h of injury, and the secondary axotomy after at least 4 h.
On the basis of their comprehensive study, Adams et al. [58] distinguished the severity of DAI. In grade 1, the damage is exclusively in the white matter and there are no focal lesions, and in grade 2, the lesion is localized to the corpus callosum. Grade 3, the most severe, is characterized by wide-spread damage to the white matter axons and local lesions in the corpus callosum and rostral brain stem. Grade 1 DAI is infrequent in VS, but occurs often in minor head injuries, serving as the pathological mechanism for clinical sequelae. The focal damage may be seen macroscopically, but is seldom observed only in microscopic examinations. In their series [58], 71 % of the brains had grade 2 or 3 DAI, with structural abnormalities, intracranial hematomas, and sustained moderate or severe ischemia. Some also had diffuse neuronal loss in the cortex, with or without “excessive pallor” of the thalamus and white matter.
In the study by Kinney et al. [57], DAI was also the more common lesion, appearing as damage to the white matter, with hemorrhagic foci in the corpus callosum and the dorsolateral quadrant of the rostral brain stem, adjacent to the superior cere-bellar peduncle. Cervos-Navarro and Lafuente [61] reported that the severity of the clinical syndrome and the outcome depend on the total number of damaged axons and their location, as well as the proportion of damaged to undamaged axons. Disruptive damage leads to axotomy, whereas nondisruptive damage is internal. Axonal repair may be attempted with both types, but is usually more successful with the second [61].
In VS, the thalamus is apparently injured via one of two mechanisms: retrograde degeneration as a result of the axonal damage, which takes several months to appear, or neuronal loss as result of ischemia, which becomes apparent a very short time after the event [58]. There is a lower frequency of damage to the cerebral cortex and/or brain stem. In the few patients affected, the lesions tend to occur in the midline, or less often laterally, in the cerebral peduncle [58]. The preservation of the hypothalamus and brain stem is characteristic of VS, as they are necessary to maintain vegetative functions. Of the 49 patients investigated by Adams et al. [58], 28 sustained damage to the thalamus. In most of them, the lesion was diffuse, and it was almost always associated with widespread damage to the white matter, with or without cerebral cortex or brain-stem damage. The importance of the thalamus was underscored by Kinney al. [57] in 1994, in their study of the brain of Karen Anne Quinlan, a 21-year-old woman who remained in VS for 10 years after a cardiopulmonary arrest. The disproportionately greater damage to the thalamus than to the cerebral cortex raised questions about the role of the thalamus in cognition and awareness. Other authors have reported similar findings [48,62].
Ischemia, brain swelling, anoxia, and acidosis play a role in the production of secondary damage after VS of traumatic origin. This includes vascular proliferation in the impact zone, which peaks in the first 3 weeks after injury and diminishes gradually thereafter over months and years, leaving functional small sclerotic vessels [61]. In the series published by Adams et al. [58], ischemic brain damage was present on arterial boundary zones in 43 % of cases, and was often accompanied by diffuse signs, indicating a global reduction in cerebral blood flow (CBS). Following the report by Graham et al. [60], they classified the ischemic damage as follows:
• Severe: diffuse or multifocal lesions or infarcts within specific arterial areas
• Moderate: ischemic damage limited to arterial boundary zones, appearing alone or in combination with subtotal infarction in the distribution of arterial territories
Ischemic lesions tend to be more severe in the presence of concomitant intracranial hematomas or extracranial injuries, particularly if the intracranial pressure is high [63].
Some researchers have postulated that over time, the volume and weight of the brain are reduced due to the striking loss of axons and neurons, so that the brain becomes more consistent, with an apparently compensatory enlargement of the ventricles [58,64]. There may also be a secondary astrocytic reaction, which increases over weeks and months, resulting in a glial scar [61]. The role of the glial proliferation is not clear. Ramón y Cajal (quoted in [61]) suggested that glia inhibit axonal regeneration, whereas Reier et al. [65] affirmed that axons can penetrate the axonal web. Other authors have supported the notion that laminin, a growth substrate for brain neurons, is produced by astrocytes following injury, inducing neuron sprouting [66–68].
Another secondary event in trauma is collagen production and fibrosis, especially when the injury occurs in the vicinity of the cortex. Fibroblasts are present around vessels, but usually disappear within a few months [61].
When the sequence of histological reactions stabilizes, a contusional zone remains, resembling an old infarction.
The same lesions seen in humans have been re-produced experimentally in primates by Gennarelli et al. [69], Jane et al. [70], and others [71,72]. The animals were subjected to high-magnitude angular acceleration of the head and long-pulsion (5–10 ms) and controlled head movements without focal loading. The severity of axonal injury depended on all three conditions and on the direction of the head motion. Sagittal acceleration produced DAI grade 1, acceleration in the coronal plane produced DAI grade 3, and horizontal acceleration produced grade 2 [53,73].
References
2. Oxford English Dictionary, 2nd ed. Simpson JA, Weiner ESC, editors. Oxford: Clarendon Press, 1989. Online: http://www.oed.com/, November 2001.
7. Webster’s Tenth New Collegiate Dictionary. Springfield, MA: Merriam-Webster, 1995.
8. Collins COBUILD English language dictionary. London: Collins, 1985.
9. Andrews K, Beaumont JG, Danze F, et al. International Working Party report on the vegetative state. London: Royal Hospital for Neurodisability, 1996 (http://www.comarecovery.org/pvs.htm).
11. Klauber MR, Barrett-Connor E, Marshall LF, Bowers SA. The epidemiology of head injury: a prospective study of entire community-San Diego County, California, 1978. Am J Epidemiol 1981; 113: 500–9.
20. Brule JF, Danze F, Vallee D. Etat végétative chronique. Rev Fr Dommage Corpor 1988; 14: 191–5.
26. Campbell AGM. Children in a persistent vegetative state. Br Med J 1984; 289: 1022–3.
31. Sazbon L. Vegetative state [personal communication].
32. Sazbon L. Prolonged coma. Progr Clin Neurosci 1985; 2: 65–81.
43. Heind JT, Laub MC. Outcome of persistent vegetative state following hypoxia or traumatic brain injury in children and adolescents. Neuropediatrica 1996; 27: 94–100.
46. Peerless SJ, Rewcastle NB. Shear injuries of the brain. Can Med Assoc J 1967; 96: 577–582.
72. Erb DE, Povlishock JT. Axonal damage in severe traumatic brain injury: an experimental study in CAT. Acta Neuropathol 1988; 76: 347–58.
< div class='tao-gold-member'>




