Anoxia, Coma, and Brain Death
Peter W. Kaplan
Gerhard Bauer
CLINICAL DEFINITIONS OF IMPAIRED RESPONSIVENESS: FROM LOCKED-IN STATES TO COMA
Consciousness
Consciousness can be described as an awareness of self and the environment. The two primary components of consciousness are awareness and arousal with the determination of consciousness being made by the examination of the patient’s response to stimuli. The examination of brainstem reflexes and responses to command is helpful in localizing the causes of coma to higher or lower CNS structures.
A further component of awareness depends on cortical integrity and subcortical connections. When these are disrupted, a patient may have a decreased consciousness to the point of being in a minimally conscious state or a vegetative state, but less frequently is in actual coma (1).
Coma
Coma is a state of unconsciousness without wakefulness, characterized by the lack of arousal from internal or external stimuli. The eyes are usually closed, and there is no awareness of self or surroundings. Brainstem reflexes may be retained. Coma is usually a transient condition that evolves to either wakefulness or death over days to weeks.
Minimally Conscious States (MCS)
Relatively recently, a minimally conscious state (MCS) has been declared a separate clinical condition (2). A MCS is that of a severely altered consciousness. Behavioral manifestations of awareness are minimal, but present. There may be fluctuating attention, as well as purposeful visual tracking and communication, but all these elements should be present. fMRI has demonstrated some patterns of brain activity in minimally conscious patients that are also seen in normal people (3). These patterns are thought to represent conscious cognitive processing.
Vegetative States (VS)
In vegetative states (VS), the patient is awake. However, there is no evidence of awareness of self or of the environment, although sleep-wake cycling occurs. There is also no evidence (during wakefulness) of cognitive processing. A VS is persistent if it has lasted at least a month, but “persistent” does not necessarily imply irreversibility. A permanent VS is irreversible, and is often diagnosed after about 3 months in nontraumatic brain injury, or a year after brain trauma. Different reports of this syndrome have used names such as apallic syndrome (4), complete and incomplete apallic syndrome, prolonged coma (5), and persistent vegetative state (PVS) (6). Absence of any clinical sign of cortical function combined with alternating periods of sleep and wakefulness is the essential of this condition. The eyes open periodically without evidence of appreciation of the environment. More recently, multimodal imaging has shed light on brain activity in the VS (1,3,7). Pathologically, no common pattern of lesion can be defined. Prognosis, EEG signs, and appropriate management rest on the strict or broadened definition of this condition (8, 9, 10 and 11). Terminological and prognostic controversies have been cleared by the Multi-Society Task Force on PVS (12). Questions remain about the boundaries to coma and VS (12).
Locked-in Syndrome (LIS)
An LIS consists of almost total paralysis of limb and head movements so that the patient cannot move or speak, but remains fully conscious (13,14). It is due to the interruption of corticospinal and corticobulbar pathways, typically in the basis pontis or midbrain (15, 16, 17, 18, 19 and 20) or from bilateral lesions in the internal capsules (21), or less frequently from total peripheral paralysis (ALS, polyradiculopathy). This pathology disconnects the brain from the ability to converse (interruption of efferent pathways). The only retained movements are of the eyelids and of vertical eye movements. The patient can communicate with a pattern of eye movements that the patient and observer have established to allow discourse. In this way, the patient demonstrates awareness and hence consciousness. EEG has shown different patterns, but the most typical is a posterior, often reactive alpha rhythm (22,23).
Akinetic Mutism
The term akinetic mutism was coined by Cairns and colleagues (1941) to describe a state of mutism, or minimal speech, and hence communication (24). There are a number of anatomical lesions that can cause this syndrome, and it is a term less used at present.
Coma Vigile
Sleep
Although sleep represents a transient absence of consciousness, a patient can be aroused to conscious wakefulness. In light coma, it can be difficult to differentiate pathologic hypersomnolence (e.g., from drugs, alcohol, or sleep deprivation) from physiologic sleepiness.
THE ANATOMICAL AND PHYSIOLOGIC SUBSTRATES OF COMA
Consciousness is maintained by the integration of ascending arousal inputs via the ascending reticular activating system through the thalami to both cerebral hemispheres. In animals and humans, these ascending pathways pass through the pons and midbrain tegmentum (26). Structural or functional impairment or interruption of these pathways in the brainstem or in both cerebral hemispheres leads to decreased consciousness and coma (27,28).
Structural Causes of Coma
The signal process needed to induce coma is compromise of brainstem function. This typically happens from compression or ischemia. Compression can arise from unilateral cerebral mass lesions exerting traction and distortion of the brainstem structures or compromise of circulation. Although bilateral cerebral hemisphere dysfunction without brainstem involvement can suppress cortical activity, coma per se rarely occurs. Further, a markedly raised ICP in itself does not induce coma, except by brainstem compromise. Pathologic examination and radiologic studies have demonstrated the effects of uncal and central herniation on diencephalic structures that mediate arousal (29), but more current imaging techniques (CT and MRI) give greatest credence to the transverse or horizontal type of herniation compromise (30,31), while evidence of downward, transtentorial herniation can be obtained with MRI (32, 33 and 34). Brainstem compromise in the latter injury arises more from anoxia than direct pressure.
More direct destruction by compression, trauma, or ischemia of the paramedian pontomesencephalic reticular formation can occur at the level of the pons and midbrain (35,36), although a direct pontine effect by trauma has been questioned (37). Other authors note rare cases of traumatic brainstem coma supported by MRI (38). Diffuse axonal injuries from torsional or shearing stresses can markedly adversely affect prognosis and be more difficult to evidence in imaging studies (39).
Toxic/Metabolic Causes of Coma
Despite the prevalence and longstanding recognition of these conditions as causes of coma, there is a relative paucity of information on their mechanism. It is believed that synaptic transmission is variably impaired, with neuronal energy failure occurring late in the process. There is early impairment of complex interactions among the cortical regions and polysynaptic pathways producing an initial impairment of higher cortical functions. In this way, judgment, memory, attention, and concentration are early heralds of impending encephalopathy and coma (40). Coma is believed to be produced by the induction of brainstem or brain edema (41, 42 and 43). The progressive impairment of consciousness mirrors, to some extent, that of anesthesia induction.
A number of mineral and electrolyte disturbances can all contribute to encephalopathy, and in the extreme, to coma. These include hyper- and hyponatremia, hyper- and hypocalcemia, hyper- and hypoglycemia, and uremia. Contributing to these electrolyte disturbances, or constituting disorders in themselves, are metabolic and endocrine disorders, and organ failures such as renal failure and dialysis effects, hypothyroidism, hypocortisolism, and porphyria. There are rarely if any pathognomonic EEG patterns with these encephalopathies and coma (44,45). The patterns reflect largely the degree of suppression (and level of coma), the presence of associated epileptiform disturbances, and occasionally focal effects. One major exception is hepatic failure that may frequently be reflected on EEG by triphasic waves (TWs) and progressive disorganization of the EEG as coma deepens even though such waves can be seen in other conditions (see later on in this chapter).
Drug intoxications in coma can produce patterns that in part reflect their cortical effects. Barbiturates progressively suppress the EEG; benzodiazepines suppress voltage, increase the power of frequencies in the beta range, and eventually slow the background. Lithium may produce epileptiform discharges, slowing, and TWs (46). Some stimulant drugs increase, voltage and frequencies, but often do not produce coma. For more detailed pathophysiology and EEG findings on toxic/metabolic and organ failure impairment of CNS function, please turn to Chapter 42.
Infection and Coma
Infection of the CNS, of individual body organs, as well as septicemia can all induce confusion and encephalopathy, but only direct CNS infection readily causes coma (47, 48 and 49). A large number of microbes may directly affect the brain and/or the meninges, along with the increasing emergence of patients with the acquired immunodeficiency syndrome (AIDS). In addition, there are conditions that may simulate infections. Conditions such as neoplasms, autoimmune conditions, and slow viruses are dealt elsewhere in this text. The pathophysiology of infection and coma has not been clearly elucidated. Mechanisms implicated include a direct release of toxins and their effect on CNS structures, the involvement of microvasculature and larger vessels, cerebral edema, a focal or generalized increase in intracranial pressure, and changes in cerebral blood flow (CBF), which may all play a part. Once again, EEG changes in these various conditions reflect the focality, epileptogenicity, level of consciousness, and the mechanism of CNS compromise produced by the respective infectious process as it induces coma.
Seizures, Epileptiform Activity, Status Epilepticus, and Coma
Epileptic activity when generalized usually causes coma. Seizures of themselves and in the form of ongoing activity (status epilepticus) generally may cause either transient or a more lasting loss of consciousness hours. Some conditions with partial brain seizure activity may impair vigilance, but do not in all cases cause coma. Coma from many causes may additionally reveal epileptiform activity and seizure activity as an “epiphenomenon.” In this sense, the underlying coma etiology is not
that of seizure activity, but rather the reflection of cortical irritability or insult. Please see also Chapters 25, 26, and 28 to 30.
that of seizure activity, but rather the reflection of cortical irritability or insult. Please see also Chapters 25, 26, and 28 to 30.
Anoxia and Coma
Because anoxia is such a common cause of coma, as well as being one of its most morbid etiologies, more attention will be paid to this entity in this chapter. The brain at rest consumes 20% of the total body oxygen consumption, even though it represents only 2% by weight (50). Consciousness and normal background EEG patterns are impaired if the brain oxygen tension falls below 15 to 20 mm Hg (51). An interruption in oxygen supply results in cell dysfunction, and ultimately death, and can be induced by a decrease in CBF, hypoxia or anoxia, a low oxygen concentration in respired air (hypoxic hypoxia). When ischemia reduces CBF below 15 mL/min/100 g of tissue for several minutes, there may be subsequent cell death and degeneration over the ensuing days, worsened by injury produced by resumed blood flow or reperfusion (52, 53 and 54). The postischemic period before cell death may represent a therapeutic window of opportunity. Causes of global ischemia include cardiorespiratory arrest (CRA), circulatory collapse, strangulation, drowning, suffocation, muscle paralysis, and toxins such as carbon monoxide. The less frequent hypoxia in pure form is rare as cardiac dysrrythmias usually also occur, as may systemic hypotension—both producing superadded ischemic damage (55). Clinical signs of anoxic-ischemic brain injury largely follow the rate and severity of the deficiency. Unconsciousness with cardiac arrest occurs in about 8 seconds in the upright position, and about 15 seconds supine, followed by tonic posturing (56). When anoxic coma is prolonged, there may be persisting deficits, with permanent cognitive decline, rigidity, ataxia and spasticity, amnesia, visual dysfunction, movement disorders, and in some cases persistent coma or a VS (57). Epileptic phenomena include myoclonus, myoclonic status, or stimulus-sensitive myoclonus (15,58, 59, 60, 61, 62 and 63). It is important to distinguish between the morbid postanoxic myoclonus in coma from the entity in which the patient returns to consciousness, but with stimulus-induced myoclonus following briefer anoxia/hypoxia (64,65). When the hypoxia is less marked, survivors are able to speak, but may have some degree of cognitive deficit. CT of the head may be normal in the early hours of the day, but will eventually reveal edema, sulcal effacement, basal ganglia lucency, and watershed changes (66,67). With MRI, there is better resolution of abnormalities in the globus pallidus, putamen, caudate nuclei, hippocampus, cerebellum, brainstem, and subcortical white matter (68, 69, 70, 71, 72 and 73). Magnetic resonance spectroscopy (MRS), diffusionweighted MRI, and positron emission tomography (PET) are all more sensitive for hypoxic insults, and at an earlier point. With PET, De Volder et al. (1990) showed hypometabolism that correlated with both location and severity of hypoxia. PET studies of patients in a VS have uncovered evidence of irreversible damage to supratentorial cortical zones (74,75).
EEG IN COMA
EEG patterns in coma may correlate with the depth of coma (76). As consciousness decreases, there is a slowing of the prevalent EEG frequencies from alpha toward delta. When there are excess theta frequencies in the posterior alpha rhythm, it may be difficult in adults to differentiate light coma from drowsiness. Occasionally, higher frequencies can be seen, for example, in the beta range, particularly with benzodiazepines and barbiturates. High doses of CNS-suppressant drugs can decrease EEG voltage. Drug effects aside, low-voltage or flatline tracings after anoxia predict an abysmal prognosis for return to consciousness.
Intermittent Delta Rhythms
Frontal intermittent rhythmic delta activity (FIRDA) (77) can be seen in mild obtundation, but rarely occurs in coma. It is believed to arise most frequently from diffuse cerebral dysfunction (78), but was initially described in deep midline dysfunctions in subcortical regions (79), deep frontal (80), and with early herniation. When FIRDA exists with slow background activity, a more diffuse cerebral dysfunction, including toxic, metabolic, or infectious etiologies, may be suspected (81,82). (see Fig. 23.1).
Prolonged Bursts of Delta Waves and the Reactivity of EEG
Some patients with head injury have paroxysms of potentiated slow activity, lasting up to minutes (83, 84, 85 and 86). These bursts may occur spontaneously or after stimulation, particularly as the patient tries to communicate.
In several of the patterns described above, spontaneous EEG variability or change in EEG patterns after stimulation indicates a more favorable prognosis. Conversely, the loss of variability or reactivity to stimuli can forebode deepening coma.
Epileptiform Morphologies
Epileptiform morphologies can be seen in many causes of coma, even without apparent clinical or electrographic seizure activity. Continuous, unilateral epileptiform discharges with little variability suggest pseudoperiodic lateralized epileptiform discharges (PLEDs), an indication of cortical irritability, believed by most authors to fall short of actual seizures. Frequent causes are structural abnormalities (e.g., strokes, trauma) with a recent seizure. Such patterns are frequently seen after seizure resolution in these settings, (87, 88, 89 and 90). When the discharges are more generalized and synchronous, usually with a relative paucity of intervening background activity, the term generalized periodic epileptiform discharges (GPEDs) has been used (91). This pattern usually reflects a more severe, diffuse cerebral insult such as is seen after anoxia (92), CNS infections, or less frequently in the end-stages of status, and correspondingly indicates a worse prognosis. Generalized discharges can be accompanied by myoclonic movements, or conversely with little evident motor manifestations (15,92,93). Bilateral independent discharges (BIPLEDs) probably lie in between with regards to both severity of etiology and prognostic significance (88). There is ongoing controversy regarding where on the ictal-interictal continuum these periodic discharge patterns lie, and hence the need (or not) of more intensive seizure suppression with anesthetic agents or anesthetic coma. Evidence that PLEDs represent a form of nonconvulsive status epilepticus (94) arises from PET studies revealing increased metabolism or blood flow (95). In most cases, parenteral AEDs fail to lastingly suppress PLEDs or improve the patient’s level of consciousness. See Figures 23.2 and 23.3.
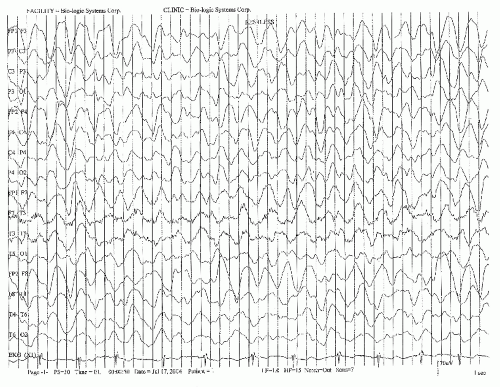 Figure 23.1 This EEG shows medium- to high-voltage theta and a predominance of arrhythmic delta activity in an unresponsive patient with multiple organ failure including renal and respiratory failure. |
Triphasic Waves (TWs)
Although this pattern was initially attributed to patients with hepatic failure and subsequently with raised serum ammonia, later case series found this pattern with several different toxic or metabolic problems, including renal failure (96). Some authors have sought to distinguish a “typical” from an “atypical” form (Figs. 23.4 and 23.5) (97,98). Typical TW pattern consist of medium- to high-voltage three-phased bilateral, symmetric waveforms in runs, with a discharge frequency of 1.5 to 2.5 Hz. Typical TWs possess an anterior-posterior phase lag, which often is not seen when using transverse or referential electrode arrays. TWs are seen with hepatic coma, after hypoxia (99, 100 and 101), and following toxic exposure (see Fig. 23.6) (2,6,96,102,103). A number of other metabolic problems have been reported with TWs (78,104). Structural lesions have occasionally been reported with TWs, including brainstem strokes and subdural hematomas (105). Infection (106) and malignancy (cerebral carcinomatosis) may also induce this pattern (107). Degenerative and spongiform encephalopathies may also produce TWs in the conscious patient (108). An important differential diagnostic consideration is the continuously occurring sharp- and slow-wave runs seen with atypical absence status in Lennox-Gastaut syndrome (109).
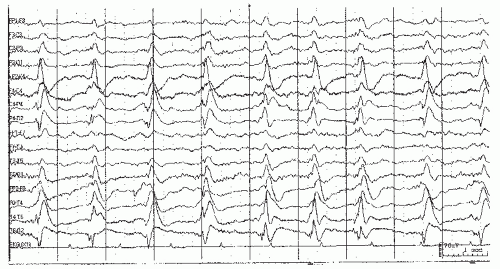 Figure 23.2. This EEG sample shows pseudoperiodic lateralized epileptiform discharges (PLEDS) at about 1 Hz over the right hemisphere in an unresponsive patient. |
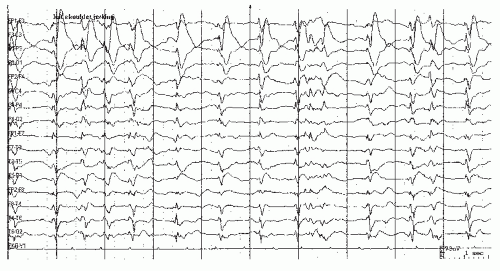 Figure 23.3 This EEG shows generalized pseudoperiodic epileptiform discharges (GPEDs) in an anoxic patient. |
Burst-Suppression Patterns
This pattern is largely associated with deeper levels of coma, and prominently occurs with anoxic insults after cardiac arrest. In this setting it carries an abysmal prognosis. Other more benign causes are in the setting of anesthetic use. In the therapeutic suppression of status epilepticus, propofol and other agents typically produce a burst-suppression pattern on the way to producing more extensive, global cerebral suppression. Overdoses or therapeutic use of barbiturates can also induce reversible patterns of burst-suppression. The pattern typically consists of high-voltage bursts of sharply contoured morphologies, and polyspikes and spikes superimposed or associated with underlying slow-wave activity. These paroxysms may last from less than a second to upwards of 10 seconds, but are characterized as burst-suppression patterns by the appearance of marked diffuse bilateral suppression of brain EEG activity between these bursts. The periods of suppression again may vary from about 1 second to periods exceeding 10 seconds. There may be a variable amount of background activity during these suppression periods (110,111). The bursts typically repeat quasi-periodically, appear diffusely and bilaterally, and exist generally throughout the recording (112). They may be associated with single or salvos of myoclonic face, arm, chest, or leg jerks (15,93,113,114). In many patients, the jerks can be induced by stimulating the patient by touch, suctioning, or loud sounds. See Figure 23.7.
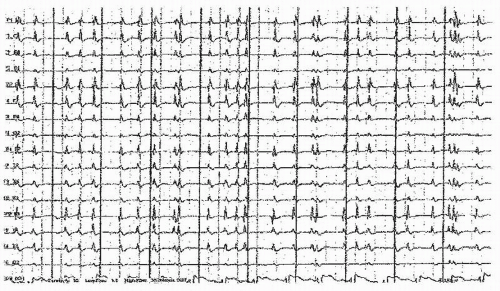 Figure 23.4 This EEG shows a pattern along the continuum of interictal to ictal, generalized periodic discharges (GPEDs) in a patient who has had a prolonged cardiorespiratory arrest, but before therapeutic hypothermia. |
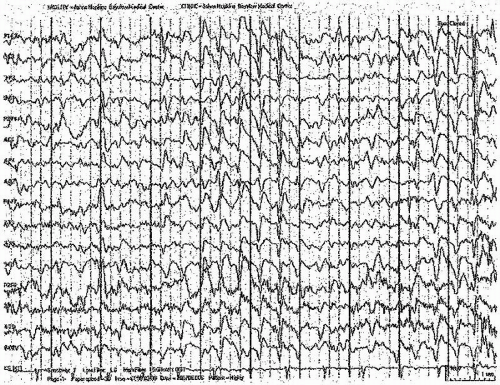 Figure 23.5 This EEG shows the effects of lithium toxicity. There are runs of triphasic waves and some sharper, epileptiform morphologies seen diffusely, bilaterally. The background is slow. The patient was obtunded during the recording, but recovered over several weeks. |
Alpha and Theta Patterns in Coma
Alpha frequency patterns may be recorded in some deep comas. Originally described with brainstem dysfunction (strokes, tumors, trauma) (115, 116, 117, 118 and 119), they were also recorded after marked anoxia (119, 120, 121, 122, 123 and 124). Differentiating alpha coma from normal, physiologic waking alpha are its diffuse and indeed frontal predilection, general lack of reactivity to noxious stimulation (or at least a failure to induce a return to discernible clinical consciousness or wakefulness), and its steady presence. Alpha coma must be distinguished from the spindle-like patterns that can also appear in unresponsive states (125,126). Drug toxicity may cause faster rhythms in the 10- to 18-Hz range (123). In the unresponsiveness of the LIS due to de-efferentation, the awake, but paralyzed patient will evince waking alpha frequencies, typically reactive to eye-opening and closure. Alpha coma may follow burst-suppression pattern (111,125), heralding death. Some studies including unresponsive patients with alpha frequencies have found that prognosis may vary according to the etiology of coma, and the presence of a degree of reactivity. Unreactive patterns and anoxia, or brainstem infarction carried the worst prognosis (see Fig. 23.8) (115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129 and 130).
Sleep-Like Patterns in Coma
Some coma EEG patterns contain spindles, slow activity, and vertex waves, resembling stage II sleep (81,131). They are usually seen after head trauma or structural lesions of the brainstem (126,132,133), but may be seen with viral encephalitis (134), and benzodiazepines (135). Such patterns may evolve to waking patterns, or diffuse slowing, and are thought to represent functional impairment of arousal mechanisms on thalamocortical circuits responsible for sleep patterns. With deepening coma, there is less variability and cycling, and the appearance of more monomorphic slow activity (see Fig. 23.9) (136).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


