Chapter 88 Anterior Horn Cell and Cranial Motor Neuron Disease
Hereditary Motor Neuron Diseases
The Anterior Horn Cells of Spinal Cord
Anterior horn cells (α-motor neurons), located in the anterior gray matter of the spinal cord, are found at every segment and are concentrated in the cervical and lumbosacral enlargements. Morphologic differentiation of the anterior horn cells is most evident from 12 to 14 weeks’ gestation [Vassilopoulos and Emery, 1977]. There is a period of normal differentiation, followed by programmed cell death [Yachnis et al., 1998; Fidzianska and Rafalowska, 2002]. Anterior horn cells are clustered into medial and lateral cell divisions (Figure 88-1). The medial group is subdivided into ventromedial and dorsomedial components. The ventromedial component innervates the superficial larger muscles, and the dorsomedial component innervates the small, deep muscles adjacent to the spine.
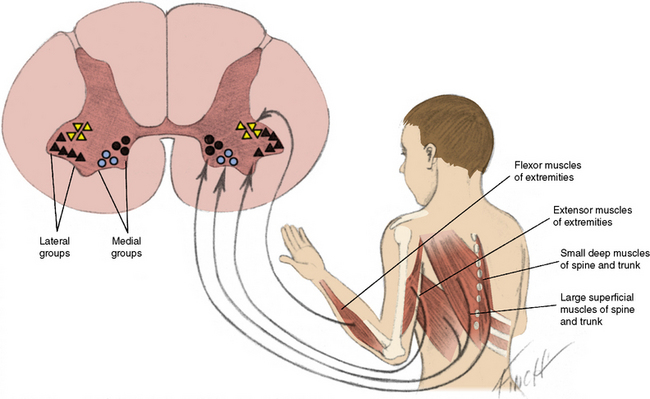
The lateral cell mass is also subdivided into groups. The ventrolateral group innervates extensor muscles, and the centrodorsal group innervates flexor muscles [Romanes, 1951]. Since these groups of neurons are located in a relatively small region, deleterious influences harm cells from more than one group and weakness may be widespread.
Autosomal-Recessive Spinal Muscular Atrophy Linked to the SMN1 Gene
Clinical Features of Spinal Muscular Atrophy
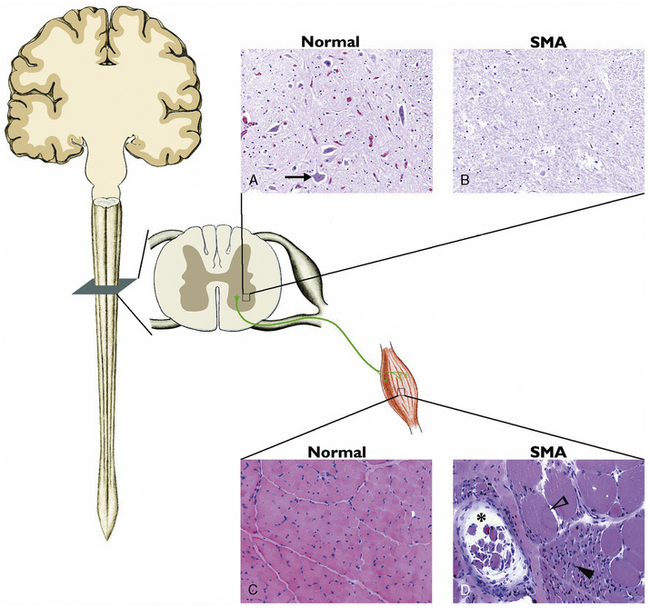
Spinal muscular atrophy (SMA) is the most common autosomal-recessive motor neuron disease of childhood, characterized by degeneration of the spinal cord and brainstem motor neurons, resulting in hypotonia and muscle weakness. Previously, SMA was diagnosed by electromyography (EMG) and muscle biopsy. EMG characteristics of SMA include spontaneous activity with positive sharp waves, fasciculation, and fibrillation. Compound muscle action potential demonstrates high amplitudes and long durations coupled with decreased recruitment. Histopathology of skeletal muscle documents atrophic fibers with islands of group hypertrophy, and the spinal cord documents severe motor neuron loss in the anterior horn region (Figure 88-2).
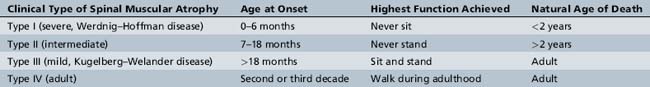
SMA is divided into three clinical types, based on age of onset and motor function achieved: the most severe type I, the intermediate type II, and the mild type III [Munsat and Davies, 1992]. Recently, an adult-onset type IV SMA was added to include very mild SMA (Table 88-1) [Russman, 2007; Wang et al., 2007].
Type i spinal muscular atrophy
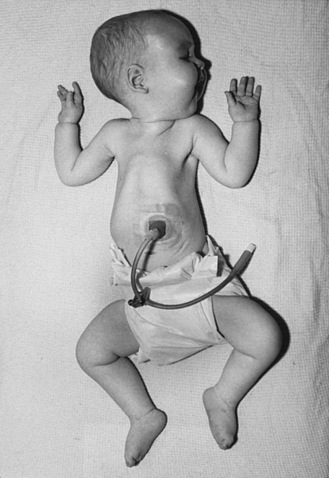
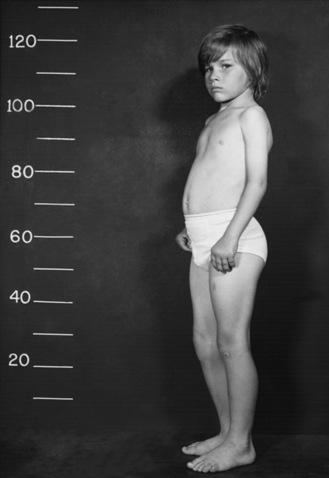
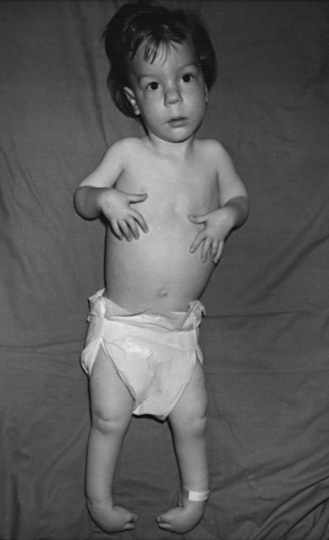
Type I SMA (Werdnig–Hoffman disease) is the most severe subtype, which accounts for approximately 50 percent of patients diagnosed with SMA [Markowitz et al., 2004]. Disease onset is usually before 6 months of age, and death occurs within the first 2 years of life. Profound hypotonia, symmetric flaccid paralysis, and poor head control are common clinical features of these patients. They are unable to sit without support (Figure 88-3). The spared diaphragm, combined with weakened intercostal muscles, results in paradoxical breathing (inward thorax movement with outward abdominal movement during inspiration) and a bell-shaped upper torso. Bulbar involvement results in tongue fasciculation and poor suck and swallow. It also leads to decreased airway protection and increased risk of aspiration pneumonia. Death is usually caused by airway complications.
Type iii spinal muscular atrophy
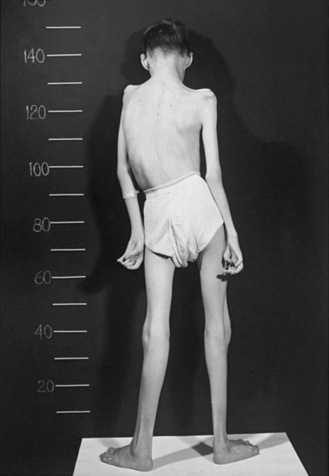
Type III SMA (Kugelberg–Welander disease) patients exhibit extreme symptom heterogeneity. These patients all achieve independent walking, but some may require wheelchair assistance in childhood, while others continue to walk and live productive adult lives with minor muscular weakness (Figure 88-4). Scoliosis often develops in these patients but later than type II SMA. Additionally, joint overuse symptoms are frequently seen.
Molecular Genetics and Pathogenesis of Spinal Muscular Atrophy
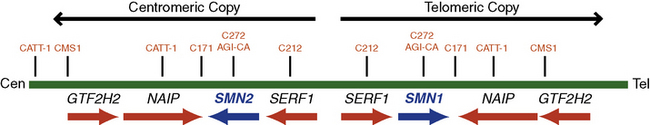
In 1990, researchers used linkage analysis to identify the SMA gene locus on chromosome 5q13 [Brzustowicz et al., 1990; Melki et al., 1990]. The initial10-centimorgan (cM) interval was later narrowed to a 1- to 2-cM region using recombinant mapping [Brzustowicz et al., 1992; Clermont et al., 1994; Wang et al., 1995]. In 1994, Melki and colleagues uncovered a small 11-kb fragment that is uniquely missing in SMA patients, thereby further narrowing the candidate genetic region. A human fetal brain complementary deoxyribonucleic acid (cDNA) library was probed by genomic DNA from this region, and the “survival motor neuron” (SMN) gene was identified as the disease-causing gene in 1995 [Lefebvre et al., 1995]. The SMN gene is present in multiple copies in the human genome: one SMN1 (SMNT, telomeric) and several SMN2 (SMNC, centromeric) (Figure 88-5). The two genes differ by only five nucleotides, two of which are within the 1.7-kb coding region, but none affects the predicted amino acid sequence [Melki et al., 1994; Schmutz et al., 2004]. Both genes contain nine exons and eight introns that span an approximately 20-kb genomic region. The SMN1 gene encodes a 38-kDa, 294-amino acid protein. The SMN protein is ubiquitously expressed in all somatic tissues and is highly conserved from yeast to humans [Lefebvre et al., 1995; Burglen et al., 1996; Schrank et al., 1997]. The SMN gene duplication found in primates occurred after the split of rodents from primates, as mice have only one copy denoted Smn. The SMN2 gene is unique to humans because nonhuman primates (e.g., chimpanzee) do not possess this gene signature, despite having multiple SMN gene copies in the genome [Rochette et al., 2001].
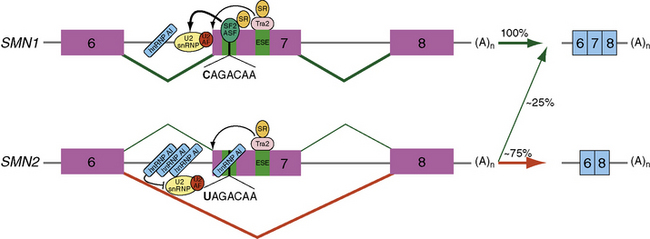
Over 98 percent of SMA patients possess a homozygous deletion or mutation of the SMN1 gene [Hahnen et al., 1995; Lefebvre et al., 1995]. All SMA patients, however, retain at least one copy of SMN2. SMN2 undergoes alternative splicing and produces a truncated messenger ribonucleic acid (mRNA) isoform lacking exon 7. A C-to-T nucleotide transition at position 6 of exon 7 (Ex7+6) is responsible for the alternatively spliced isoform lacking exon 7 (Figure 88-6) [Lorson et al., 1999; Lorson and Androphy, 2000]. The resultant SMNΔ7 protein is nonfunctional and thought to be rapidly degraded [Lorson and Androphy, 2000; Chang et al., 2004; Vitte et al., 2007]. Approximately 25 percent of SMN2 pre-mRNA is properly spliced and subsequently translated into full-length SMN protein. This low level of SMN protein permits embryonic development but is not sufficient to sustain the survival of spinal cord motor neurons; thus, the occurrence of SMA.
Cellular Function of SMN Protein
SMN protein is expressed in the cytoplasm and nucleus in all somatic cells, with high levels in motor neurons of the spinal cord [Battaglia et al., 1997; Coovert et al., 1997]. Within the cytoplasm, SMN protein localizes to several small discrete structures associated with coiled (Cajal) bodies named “gems” (“gemini of coiled bodies”) [Liu and Dreyfuss, 1996; Carvalho et al., 1999]. While the exact cellular function of gems remains unknown, cells from SMA patients contain significantly fewer gems compared with those in gene carriers and non-SMA controls [Coovert et al., 1997; Grzeschik et al., 2005]. In eukaryotic cells, the spliceosome is the cellular organelle that executes post-transcriptional pre-mRNA splicing. Uridine-rich small nuclear ribonuclear proteins (U snRNPs), the principal components of spliceosomes, recognize highly conserved sequences and ligate exons together. SMN protein, part of a highly stable complex with at least eight other proteins (reviewed in Otter et al. [2007]), is necessary and sufficient for proper assembly of Sm (Smith class) core proteins in the U snRNP [Meister et al., 2001; Pellizzoni et al., 2002]. A point mutation (E134K) in the SMN tudor domain, the region that mediates Sm protein assembly [Buhler et al., 1999], can lead to SMA disease by affecting the charge distribution in the SMN-Sm binding site.
While SMN protein is ubiquitously expressed in all somatic cells, it is puzzling why the spinal cord motor neurons are specifically vulnerable in SMA. Recent studies suggest that SMN protein may play an important role in certain cellular functions that are unique to motor neurons. SMN protein was recently found to localize in RNP granules in the neurites and growth cones of primary motor neurons and in embryonic stem cell-derived motor neurons [Fan and Simard, 2002; Zhang et al., 2006]. Active bidirectional transport of these granules to neuronal processes and growth cones was observed by live-cell fluorescence microscopy [Zhang et al., 2006]. SMN protein also interacts with heterogeneous nuclear ribonucleoprotein (hnRNP) R and Q [Mourelatos et al., 2001; Rossoll et al., 2003]. The former interacts with the 3′ untranslated region (UTR) of β-actin mRNA [Rossoll et al., 2002]. Low levels of SMN protein resulted in low β-actin mRNA and protein levels in axons and growth cones [Rossoll et al., 2003], further suggesting that SMN protein may be involved in transportation of RNP complexes containing β-actin. Beta-actin deficiency results in motor neuron axon outgrowth and pathfinding defects in two neuronal culture models [Zhang et al., 2003] and in a zebrafish model of SMA [McWhorter et al., 2003]. Profilin, an actin-binding protein, regulates actin dynamics through sequestration and release of actin monomers. SMN protein co-localizes with profilin IIa (the predominant isoform in neuronal tissue [Lambrechts et al., 2000]) in distinct granules in neurite-like extensions and growth cones. Mutant SMN protein fails to interact with profilin IIa, and knocking down profilin IIa alone inhibits neurite outgrowth [Sharma et al., 2005].
Other Hereditary Diseases Affecting Spinal Motor Neurons
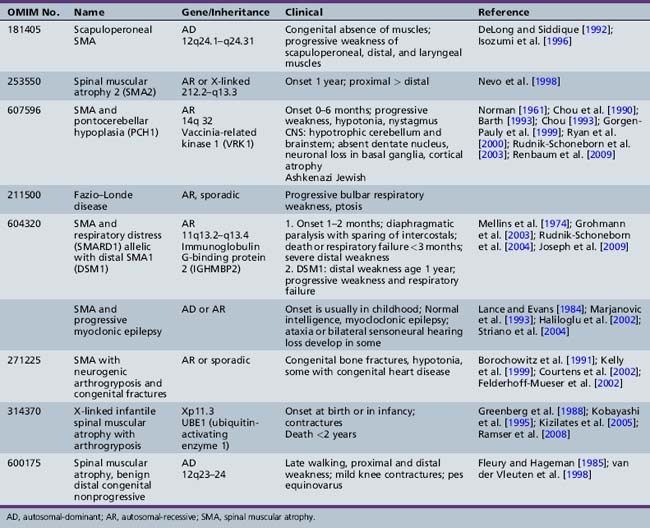
Other children with proximal weakness onset at birth or within the first year of life also manifest anatomic and electrodiagnostic evidence of motor neuron disease not linked to the SMN gene [Guillot et al., 2008]. Additional or atypical features are present in these children. Several of these conditions are now well characterized and genes have been identified (Table 88-2). Pontocerebellar hypoplasia (PCH1) with SMA was first described by Norman in an infant who experienced cognitive decline and died at 6 months [Norman, 1961]. The gene responsible for this autosomal-recessive disease was subsequently mapped to 14q32. In 2009, the responsible gene (vaccinia-related kinase 1) was determined by Renbaum et al. [Renbaum et al., 2009]. Scapuloperoneal SMA was first described clinically by Emery prior to the identification of the SMN gene, and Mawatari recognized sporadic and X-linked cases [Emery et al., 1968; Mawatari and Katayama, 1973]. More recently, a clinically similar syndrome with onset in the first decade of life and slow progression has been linked to chromosome 12 with autosomal-dominant inheritance [DeLong and Siddique, 1992; Isozumi et al., 1996].
Table 88-2 Rare and Atypical Forms of Proximal or Bulbar Spinal Muscular Atrophy Not Linked to the SMN Gene
Arthrogryposis Multiplex Congenita and Motor Neuron Diseases
Arthrogryposis multiplex congenita, a syndrome manifesting at birth, is characterized by multiple, fixed contractures of large joints. The pathologic features of arthrogryposis cover a broad spectrum [Banker, 1985, 1986, 1994]. General and genetic classifications have been proposed [Hall, 1997]. Fetal hypokinesia or restricted movement in utero of any cause will result in congenital contractures. Congenital anomalies of the brain are common in this group of children (e.g., hydrocephalus, hydranencephaly, microcephaly) [Hageman et al., 1987; Hennekam et al., 1991], and may be accompanied by anomalies of other organs [Vlaanderen et al., 1991]. Inheritance of anterior horn cell arthrogryposis, when determinable, is most often autosomal-recessive. However, X-linked and autosomal-dominant modes of transmission have been reported [Fleury and Hageman, 1985].
Anterior horn cell arthrogryposis results from a marked decrease of the anterior horn cell population in the cervical and lumbar spinal cord enlargements. More than 90 percent of patients with arthrogryposis have anterior horn cell-related disease [Banker, 1985; Adams et al., 1988]. Most affected infants hold their arms rotated inwardly; elbow position varies. The forearms are held pronated, with ulnar deviation at the wrists. The hands are flexed, and the fingers are curled tightly. The thighs are usually externally rotated and flexed at the hips. There is either flexion or extension of the knees, but the feet are almost always positioned in an equinovarus deformity (Figure 88-7). The limbs are slender (Figure 88-8); conversely, the joints appear greatly enlarged (Figure 88-9). The patellae may be very small or absent. Other abnormalities may include inguinal hernias, cleft palate, scoliosis, ankylosis of the temporomandibular joint, and cardiac malformations [Drachman and Banker, 1961].
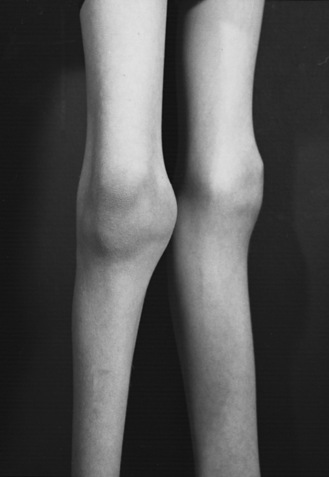
Passive movement of the affected joints is possible, but only a small range of motion is present. Deep tendon reflexes are not elicitable, and electromyographic examination confirms electrical abnormalities consistent with anterior horn cell impairment. The available pathologic studies of the involved muscles revealed markedly decreased bulk [Drachman and Banker, 1961] or even absence [Adams et al., 1988]. Hip contractures develop from unopposed contraction of the iliopsoas muscles. Muscle spindles are normal. Few pathologic alterations are evident in endomysial connective tissue, but perimysial connective tissue may be decidedly increased. One affected member each in two sets of identical twins has been reported [Drachman and Banker, 1961]. Children with arthrogryposis associated with anterior horn cell disease present in infancy may demonstrate autosomal-recessive, sporadic, or X-linked inheritance. The genetically defined syndromes are described in Table 88-2. Pathological features of arthrogryposis multiplex congenita associated with anterior horn cell dysfunction include infants from two families who had flexed elbows, extended wrists, acute flexion of the knee and hip joints, and bilateral pes equinovarus [Pena et al., 1968; Hooshmand et al., 1971]. Postmortem examination documented nodular fibrosis of the anterior spinal roots along the entire length of the spinal cord and absence of myelin and axis cylinders. Central chromatolysis was evident in a few anterior cells; the morphologic changes were believed to be secondary to the anterior root lesions. The skeletal muscle displayed neurogenic muscular atrophy. Identification of muscle tissue in some areas was impossible because of extensive replacement by fat [Pena et al., 1968].
A rare form of lethal arthrogryposis multiplex congenita related to abnormal Schwann cell development has been reported [Charnas et al., 1988]. Contractures have accompanied neonatal myasthenia gravis, although anticholinesterase therapy does not improve them [Holmes et al., 1980; Smit and Barth, 1980].
Prenatal diagnosis using ultrasound [Stoll et al., 1991; Bonilla-Musoles et al., 2002] is now common. Newborns with arthrogryposis may also have myotonic dystrophy [Sarnat et al., 1976]. The lower extremities are predominantly involved. These infants often have loose, wrinkled skin and a paucity of subcutaneous tissue. Neck muscle weakness precludes normal neck flexion. Serum creatine kinase activity may be elevated. Diagnosis can be confirmed by blood DNA analysis.
Considering the differential diagnosis, it is not surprising that muscle pathology varies greatly in arthrogryposis patients. All of the congenital myopathies have been associated with arthrogryposis, including central core disease, nemaline myopathy, and centronuclear myopathy, as well as congenital muscular dystrophy [Moerman et al., 1985; Schmalbruch et al., 1987; Quinn et al., 1991]. Selective distal arthrogryposis is more likely to be caused by specific muscle mutations [Sung et al., 2003a, b]. Administration of anesthesia during surgical intervention may be complicated by malignant hyperthermia, especially in children with muscular dystrophy or central core disease [Baines et al., 1986].
Arthrogryposis has been reported in Alaskan Eskimos, named Kuskokwim disease for the river delta on which the original cases were diagnosed [Petajan et al., 1969]. Multiple contractures of the joints, commonly of the knees and ankles, characterize the disease. The primary pathologic process is associated with the connective tissue. There is no indication of myopathy, neuropathy, or motor cell disease. The disease manifests in the first decade of life and is transmitted as an autosomal-recessive trait. Contractures at the elbows and knees may be seen during the first few months of life. Accompanying internal tibial torsion and profound planovalgus occur. Flexion hip contractures are also present. The deep tendon reflexes are usually elicitable in the biceps and Achilles tendons, but are frequently absent in the triceps and quadriceps tendons. Although one infant had a small cataract at 3 years of age, cataracts are uncommon in adults. Radiographic examinations have revealed cysts in the proximal long bones of several patients. Other patients have had pigmented nevi and diminished corneal reflexes. This form of arthrogryposis may be associated with webbed-neck or Klippel–Feil deformity, and with anomalies of the genitourinary system [Beckerman and Buchino, 1978].
New information describing genes that control development of limb buds suggests that abnormalities in transcription factors may be theoretically responsible for some cases of arthrogryposis [Roberts and Tabin, 1994; Nelson et al., 1996]. In such cases, there would be no evidence of muscle or nerve disease but muscle development in the limbs may be severely defective or absent (amyoplasia) [Sells et al., 1996]. To date, no such mutations have been found. On the other hand, mutations in contractile proteins, such as tropomyosin 2 and troponin I, have been documented to cause amyoplasia or distal arthrogryposis [Sung et al., 2003a, b]. Mutations in HOXD10 and homeobox PITX1 have also been described in isolated distal vertical talus [Dobbs et al., 2006] and clubfoot, respectively [Gurnett et al., 2008].
Orthopedic procedures performed on patients with arthrogryposis often provided sufficient alignment and stability of the legs to allow independent ambulation [Fisher et al., 1970; Bennett et al., 1985; Guidera and Drennan, 1985]. Talectomy before walking ability is acquired may prove beneficial [Solund et al., 1991]. Scoliosis may be a prominent problem and may require surgical correction [Daher et al., 1985]. Some improvement in arm and hand function is usually possible with physical therapy and exercise. Anesthesia for these patients requires special consideration [Baines et al., 1986; Oberoi et al., 1987]. Otolaryngologic management may be necessary for poor suck reflex, omega-shaped epiglottis, airway compromise, achalasia, and micrognathia [Laureano and Rybak, 1990].
Motor Neuron Diseases Involving Bulbar Functions
In progressive bulbar paralysis of childhood, Fazio–Londe disease, motor neurons in cranial nerve nuclei may be progressively impaired and decreased in number, with resultant progressive bulbar paralysis and little or no accompanying anterior spinal cord involvement [Gomez et al., 1962]. While the condition may be genetically transmitted [Fazio, 1892; Londe, 1893, 1894], sporadic cases are more common [McShane et al., 1992; Suresh and Deepa, 2004] and there has been no gene identified. Age of onset varies and may be as early as 3 years. Bulbar paralysis is only one facet of progressive motor neuron disease in many of these patients, and pathologic study confirms widespread degenerative changes in the brainstem. Morphologic involvement of anterior horn cells has been demonstrated, despite the absence of clinical signs [Alexander et al., 1976]. Involvement of the anterior horn cells in the cervical and upper thoracic cord segments is often associated with demonstrable loss of neurons in other locations, including decreased neuronal population in the dentate nucleus. Of the cranial nerves, cranial nerve VII is almost always affected. Cranial nerve XII is usually affected pathologically, and clinical manifestations are apparent in the early-onset patients. The nuclei of cranial nerves III, IV, VI, and X may also be involved. Clinical impairment of extraocular movement is rare.
Juvenile-onset bulbospinal muscular atrophy associated with deafness (Brown–Vialetto–Van Laere syndrome) manifests late in the first decade or in the second decade [Brown, 1894; Vialetto, 1936; Van Laere, 1966]. Bulbar impairment is indicated by dysarthria, dysphagia, and facial diplegia [Gallai et al., 1981; Summers et al., 1987].
Spinal and bulbar muscular atrophy (kennedy disease)
Kennedy disease or spinal and bulbar muscular atrophy (SBMA) was first described in 1968 [Kennedy et al., 1968]. It is a rare X-linked motor neuron disease that causes proximal weakness, bulbar weakness with dysphagia and aspiration, asymmetric or symmetric facial weakness, and gynecomastia. Age of onset is variable, and diagnosis may not be possible until gynecomastia develops after the fourth decade [Atsuta et al., 2006]. Fischbeck et al. localized the gene in seven families to Xq12–22 [Fischbeck et al., 1986]. Subsequently, the human androgenic receptor gene (hAR) was localized to Xq11–12, and examination of hAR in SBMA patients demonstrated that there was an abnormal expansion of exon 1 to more than twice normal size [Choi et al., 1993]. The expansion was found to be caused by CAG repeats, normally numbering 15–31 in hAR, but numbering more than 40 in the SBMA mutation [La Spada et al., 1991; Belsham et al., 1992; La Spada et al., 1994].
While the age of genetic diagnosis is usually in the fourth of fifth decade of life when weakness is present, the earliest symptoms may be present in the second decade of life and include gynecomastia, muscle cramps, and fatigue [Sperfeld et al., 2002]. This weakness is slowly progressive and involves bulbar musculature, including a weak, atrophic tongue. There are no signs of spinal cortical tract dysfunction; deep tendon reflexes are diminished or normal. Fasciculations and cramps in limb muscles are common. Hypesthesia and decreased vibratory sense are present in about half of cases. There may be mild elevation of muscle enzymes, but electrodiagnostic study and muscle biopsy are indicative of motor neuron disease. Electrodiagnostic studies indicate that motor involvement correlates directly with CAG repeat length, but sensory involvement corresponds inversely with CAG repeat length [Suzuki et al., 2008]. Once the clinical phenotype is recognized in a patient, diagnosis may be confirmed by mutation analysis in a blood sample.
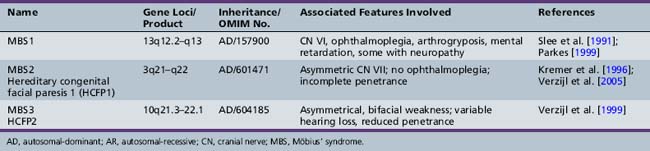
Möbius’ syndrome
First described in 1898 by Thomas, Möbius’ syndrome is a congenital, static condition manifested by facial palsy (often bilateral) and involvement (usually bilateral) of cranial nerve VI [Thomas, 1898]. The lower face is usually less involved than the upper face. Impaired lateral rectus muscle contraction results in unopposed medial rectus muscle action, with resultant convergence (Figure 88-10). Conjugate eye movements in the vertical plane are usually present, although ptosis may also occur. Dysfunction of other cranial nerves, including cranial nerves V, X, XI, and XII, often accompanies the abnormalities of cranial nerves VI and VII. Associated absence of the pectoral muscles may occur. The clinical findings are correlated with failure of development or degeneration of the involved cranial nerve motor cells. When three different gene loci were identified, there was a change in nomenclature to reflect this (Möbius’ syndromes 1, 2, and 3 (Table 88-3). However, additional pathological studies suggested that Möbius’ syndrome 1 demonstrates more hypoplasia of the entire brainstem. Thus, the nomenclature concurrently used often refers to Möbius types 2 and 3 as hereditary facial nerve palsy (HFNP) types 1 and 2 [Verzijl et al., 2005]. Acquired Möbius’ syndrome has also been associated with exposure to cocaine or misoprol in utero [Kankirawatana et al., 1993; Shepard, 1995; Leong and Ashwell, 1997; Pastuszak et al., 1998; de Muelenaere, 1999; Vargas et al., 2000; Goldberg et al., 2001; Marques-Dias et al., 2003]. While the differential diagnosis includes non-motor neuron involvement, such as supranuclear or myopathic conditions like facial scapulohumeral dystrophy, myotubular myopathy, or myotonic dystrophy, other clinical features usually will distinguish Möbius’ syndrome from these.
Amyotrophic lateral sclerosis
While most ALS occurs in a sporadic fashion, multiple genes or gene loci have now been identified in familial ALS, which is present in about 10 percent of all patients (Table 88-4). In 1991, Siddique et al., in international collaboration, reported the linkage of one form of familial amyotrophic lateral sclerosis to 21q22.1–22.2 in 23 families. SOD, a gene that encodes a cytosolic Cu/Zn-binding superoxide dismutase, is causative in ALS1 [Deng et al., 1993; Rosen et al., 1993]. The role of superoxide dismutase in controlling free radical tissue levels made it a logical choice as a candidate gene in familial ALS. Superoxide is known to be cytotoxic, but the accumulation of superoxide by an inhibition of superoxide dismutase may be exacerbated by activation of excitatory neurotransmitters, such as glutamate [Coyle and Puttfarcken, 1993]. Transgenic mouse models have helped understanding of novel specific pathogenic mechanisms. While SOD1 knockout mouse has no weakness, severity of weakness in this model is related to transgene copy number. Mice with high copy numbers develop progressive weakness and motor neuron loss, resulting in death [Borchelt et al., 1998; Shibata, 2001].
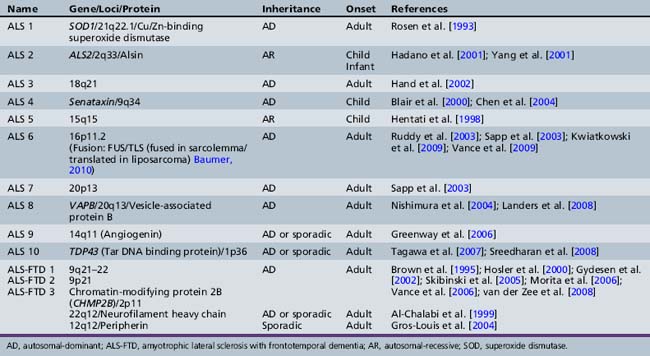
In addition to ALS 1 caused by SOD, other specific genes or gene linkages are associated with adult-onset disease (see Table 88-4). Sporadic and familial types of ALS are clinically indistinguishable. Familial ALS has been studied to understand pathogenesis. There are three well-recognized ALS syndromes with onset in the first two decades of life. ALS 2 is an autosomal-recessive disease due to mutations in the Alsin gene [Hadano et al., 2001; Yang et al., 2001]. Alsin is a member of the guanine nucleotide exchange factors for the small guanosine triphosphatase, RAB5, and plays a role in endosomal trafficking [Hadano et al., 2006]. Children present in the first decade of life with spasticity involving face and limbs, and may develop pseudobulbar affects. Symptoms are very slowly progressive, with difficulty walking after age 40. An allelic condition, progressive lateral sclerosis with onset in infancy, has also been found to be associated with mutations in this gene [Eymard-Pierre et al., 2002; Gros-Louis et al., 2003; Verschuuren-Bemelmans et al., 2008]. Alsin-deficient mice experience progressive axonal degeneration [Yamanaka et al., 2006].
The second juvenile-onset form of ALS is ALS 4, an autosomal-dominantly inherited distal motor neuronopathy with pyramidal symptoms [Chance et al., 1998]. Symptoms begin in the second decade of life with difficulty walking, and examination indicates both upper and lower motor neuron involvement. Bulbar symptoms are infrequent. Electrophysiology confirms localization to the level of the anterior horn cell [Rabin et al., 1999]. The clinical course is one of slow progression, with use of wheelchair by fifth or sixth decade and a normal life span. Recently, causative mutations in the Senataxin gene (SETX) have been demonstrated. While the precise function is unknown, the senataxin gene has homology to genes involved in RNA processing [Chen et al., 2004]. Allelic variants include autosomal-recessive ataxia oculomotor apraxia type 2 [Moreira et al., 2004]. The third form of ALS with childhood onset is ALS 5, localized to chromosome 15q15.1–q21.1; this type is also recessively inherited. No specific gene has been identified. This form of juvenile ALS has been described in multiple ethnic groups from North Africa, South Asia, and Europe. There are two clinical subtypes. Onset for both is in the second decade of life. Type 1 manifests arms more involved than legs and bulbar symptoms developing late. Type 2 is characterized by much more spasticity and much more prominent involvement of the legs, and may be a form of familial spastic paraparesis [Hentati et al., 1998].
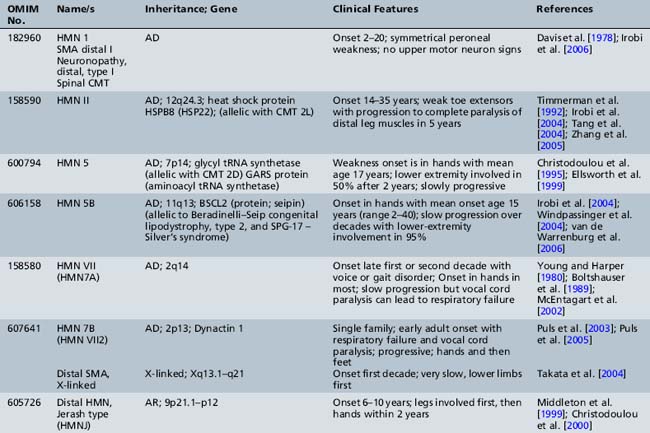
Distal Spinal Muscular Atrophy and Hereditary Motor Neuropathy
Informative families and molecular genetics have allowed researchers to identify multiple distal forms of SMA (neuronopathy) that may be difficult to distinguish clinically from hereditary motor neuropathies. Since positional cloning has been developed, several disease-associated genes have been identified for autosomal-dominant, autosomal-recessive, and X-linked distal hereditary motor neuropathy (reviewed by Irobi et al. [Irobi et al., 2006]). Chromosomal linkage is present for many others (Table 88-5). Many of these identified genes code for housekeeping functions, including RNA processing, apoptosis, and stress response, suggesting that the motor neuron is particularly vulnerable to these defects [Irobi et al., 2006]. Localization to the motor neuron is suspected when the clinical picture demonstrates distal weakness and atrophy with normal sensation. It is confirmed with electrodiagnostic studies that show normal or near-normal sensory studies, normal conduction velocities, but chronic denervation on electromyography testing.
Motor Neuron Diseases of Unknown Etiology
Monomelic Amyotrophy (Hirayama’s Disease)
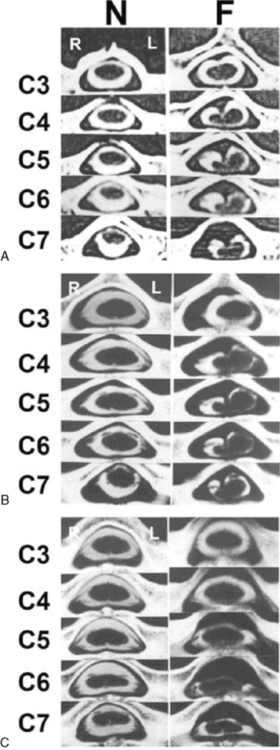
In 1959, Hirayama et al. described a form of monomelic atrophy in young Japanese patients [Hirayama et al., 1963]. This disorder has also been called O’Sullivan–McLeod syndrome for description of similar patients several years later [O’Sullivan and McLeod, 1977]. This form of sporadic disease occurs predominantly in young men (10:1 ratio) and involves the hand and forearm. While it is usually unilateral, it can be bilateral, with progressive weakness over 1–4 years, followed by a plateau. A few familial cases have been described [Schlegel et al., 1987; Tandan et al., 1990; Nalini et al., 2004] but no causative gene is known. The precise pathogenesis is not known but recent imaging evidence suggests neck flexion may induce cervical myelopathy (Figure 88-11), and pathology of the dura mater obtained at surgery suggests that elastic fibers are decreased in patients [Yoshiyama et al., 2010].
Benign Congenital Hypotonia
When tone is examined in infants and young children, both axillary and appendicular tone should be assessed. Infants with central nervous system injury often demonstrate decreased axial tone but increased appendicular tone after the first months of life. However, in the first months, tone in both areas may be decreased in these infants. In contrast, children with hypotonia secondary to disorders of spinal cord (SMA), nerve, neuromuscular junction, or myopathy usually maintain low axial and appendicular tone and have clear weakness throughout infancy and childhood. The initial reports of children with “benign congenital hypotonia” cited 17 infants with hypotonia at birth or in the first months of life. Eight improved completely by 10 years, and the other 9 improved but had definite muscle weakness or severe ligamentous laxity. All 8 children who recovered had deep tendon reflexes when initially examined, although 3 had reflexes that were difficult to elicit [Walton, 1957]. While, there was no clinical evidence of muscular dystrophy, infantile spinal muscular atrophy, mental deficiency, or upper motor neuron disease in these children, diagnostic studies were limited. Just 2 had muscle biopsies with hematoxylin and eosin staining only [Walton, 1957]. Children with “benign hypotonia” may have various conditions, including nonprogressive congenital myopathies identified with more detailed testing [Spagnoli et al., 1985; Thompson, 2002]. Some of these disorders have clinical or hereditary implications, making these specific diagnoses important. Other children with hypotonia in infancy without weakness may have cognitive difficulties later [Dubowitz, 1968; Zellweger, 1983; Shuper et al., 1987]. Thus, some suggest that this diagnosis be eliminated or be replaced with the term central hypotonia because the prognosis is not necessarily benign. The cause in those children without a neuromuscular diagnosis is likely related to improper modulatory influences on the gamma loop from the central nervous system. As the brain matures, typical motor deficits may develop that are consistent with a diagnosis of cerebral palsy. The condition may be accompanied by cognitive deficit [Dubowitz, 1968]. In short-term follow-up to age 3 years, 44 percent of 36 children still had hypotonia and gross motor delay, and a small number had speech delay [Shuper et al., 1987]. One recent follow-up of 25 children with a clinical diagnosis of benign congenital hypotonia demonstrated that, by age 6–8 years, these children still had inferior gross motor performance for coordination and strength, compared to 26 healthy controls [Parush et al., 1998]. Thus, the clinician must be wary of making the diagnosis of benign congenital hypotonia because of the large differential diagnosis, which includes infantile spinal muscular atrophy and congenital myopathies [Zellweger, 1983; Thompson, 2002]. Follow-up examination to determine if the appendicular tone increases or stays low is critical to deciding which children should have more detailed diagnostic studies [Thompson, 2002]. It is also possible that some children with congenital hypotonia without persistent weakness have variable degrees of ligament laxity [McGrory et al., 1997; Carboni et al., 2002].
Spinal Cord Anomalies Affecting Motor Neurons
Syringomyelia
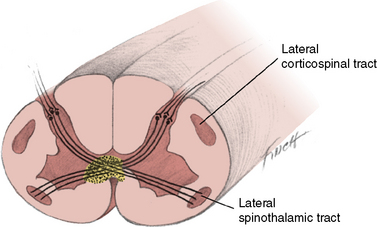
Syringomyelia is a slowly progressive cavity formation within the spinal cord, medulla oblongata, or both, which is associated with gliosis. When the medulla is involved, the condition is called syringobulbia. Cavity formation usually occurs in the cervical and lumbar cord segments. On cross-section, the destruction occurs in the area of the anterior white commissure, interrupting crossing fibers of the lateral spinothalamic tracts, with resultant bilateral loss of temperature and pain sensation (Figure 88-12). Superficial sensation is normal because fiber tracts associated with this function are located dorsally and do not cross the cord. Dissociation of sensation occurs when there is concurrent preservation of light touch perception and decrease or absence of pain and temperature perception. The anterior horn cells are affected only after the lesion has reached significant proportions.
Chiari I malformations are often accompanied by syringomyelia or hydromyelia [Gillespie et al., 1986; Isu et al., 1990; Isu et al., 1992; Inoue et al., 2003]. The association of Chiari I malformation with syringomyelia is poorly understood [Gillespie et al., 1986; Iqbal et al., 1992]. After surgery for Chiari malformation, syringomyelia may persist [Tubbs et al., 2004]. In one set of monozygotic twins, both had syringomyelia but they were discordant for Chiari I malformation [Murphy et al., 2006]. Other congenital structural abnormalities, including basilar impression, Klippel–Feil syndrome, Dandy–Walker malformation, cervical ribs, and scoliosis, may be associated with syringomyelia [Berke and Magee, 1976; Isu et al., 1990, 1992; Jaeger et al., 1997; Kasliwal et al., 2008]. Trauma may also cause syringomyelia [Reed et al., 2005]. Growth hormone deficiency is also recognized to be associated with syringomyelia [Murphy et al., 2007] and resolution may occur with growth hormone replacement [Gupta et al., 2008].
The earliest clinical presentation of syringomyelia may be isolated congenital scoliosis or kyphosis. Magnetic resonance imaging (MRI) studies of these infants indicate that 20–30 percent have associated spinal cord abnormalities, including syringomyelia, diastematomyelia, and tethered cord [Suh et al., 2001; Dobbs et al., 2002].
Incomplete closure of the neural tube during the fourth week of gestation is the likely cause of congenital syringomyelia. The normal pattern of cell differentiation causes an inner clustering of spongioblastic cells, which subsequently form glial tissue, and an outer clustering of neuroblastic cells, which subsequently form neurons and their processes. Congenital syringomyelia may result from delayed differentiation of the spongioblastic cells, with ensuing cavitation and gliosis [Leyden, 1876].
Another theory cites a possible congenital structural defect of the hindbrain reminiscent of the one that leads to myelomeningocele and the Chiari deformity. Gardner reported many instances in which the foramen of Magendie was sealed by a membrane [Gardner, 1960]. Clear fluid appeared to accumulate between the syrinx and the fourth ventricle. Failure of the foramen to form an adequate passageway for the fluid during gestation may cause distention of the syrinx and fourth ventricle. Although these findings occasionally may reflect hydromyelia, it is more likely that the pathophysiologic defect is an abnormal intramedullary vascular pattern. The vessels appear to be susceptible to occlusion; thus, infarction or hemorrhage is likely to occur and cause cavitation [Netsky, 1957]. This vascular compromise may come as a result of intermittent compression from increased pressure [Levine, 2004; Brodbelt et al., 2003a].
Another explanation for syringomyelia evolved from a questionnaire designed to determine whether a high number of mothers of children with syringomyelia had experienced difficult labor [Williams, 1977]. An unexpectedly high number of patients had undergone forceps delivery. The number of instances in which the patient with syringomyelia was the firstborn was also significantly increased. It was suggested that, as a result of birth injury, the tonsils descend through the foramen magnum and cause arachnoiditis. This mechanism may only be relevant for infants who already have a Chiari malformation [Newman et al., 1981]. The difference in fluid pressure between cranial and spinal compartments may lead to on-going insidious descent of the tonsils, resulting in communicating syringomyelia.
Recent animal models to study syringomyelia have focused on post-traumatic models. These models have very high syrinx formation when an excitotoxic agent is added to the traumatic injury. In the rat model, fluid enters the syrinx from the subarachnoid space via perivascular spaces. There is more flow in the perivascular spaces at the levels of the syrinx [Yang et al., 2001; Brodbelt et al., 2003a, b, c].
Patients with syringomyelia have limited abilities to monitor pain and temperature. Trophic ulcers of the fingers are often present. Weakness of the limbs is often profound because the cervical and lumbosacral enlargements bear the brunt of the cavitation and gliosis. Muscle atrophy is often first appreciated in the distal small hand muscles. Deep tendon reflexes are either difficult to elicit or absent. Fasciculations are visible in muscles of the shoulder, elbow, and hip. Cervical syringomyelia has been accompanied by papilledema. Elevated intracranial pressure may be present and results from hydrocephalus, mass lesions, or obstruction of the foramina of Luschka and Magendie [Alpers and Comroe, 1931]. Rarely, syringomyelia may be acquired from tumors, or infections of the spinal cord. These acquired types are unusual in infants and young children. Trauma may be a more common cause of acquired syringomyelia in older children and adults [Schurch et al., 1996; Silver, 2001].
Intramedullary cord tumors may cause signs and symptoms similar to those of syringomyelia and prove confounding [Williams, 1977; Gillespie et al., 1986; Madsen et al., 1994]. Differentiation between syringomyelia and intramedullary tumors is possible with modern imaging techniques.
Spinal fluid dynamics and constituents are usually normal in syringomyelia, although the fluid may be xanthochromic and flow may be obstructed. The Valsalva maneuver, which accompanies coughing, straining, and postural changes, may increase pressure and grossly exaggerate findings associated with cysts in the cord. The transient increased pressure may enhance the formation of syringomyelia and syringobulbia [Bertrand, 1973; Levine, 2004].
MRI appears to be the most sensitive method in the detection of syringomyelia [Gillespie et al., 1986; Iqbal et al., 1992]. Management of syringomyelia accompanied by Chiari I malformation consists of posterior fossa decompression, which is beneficial in 50–75 percent of such patients [Vaquero et al., 1990; Attenello et al., 2008]. Motor symptoms may improve more than sensory symptoms, and clinical improvement precedes radiographic improvement [Attenello et al., 2008]. Iskandar et al. demonstrated normalization of cerebrospinal fluid flow postoperatively in children with Chiari I malformation, with or without associated syringomyelia [Iskandar et al., 2004]. Children with syringobulbia also benefit significantly from posterior fossa decompression [Greenlee et al., 2005].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


