Chapter 27 Hydrocephalus and Arachnoid Cysts
Hydrocephalus
Definition
More recently, hydrocephalus has been more broadly defined as a disturbance of formation, flow, or absorption of CSF that leads to an increase in volume occupied by this fluid in the central nervous system (CNS) [Rekate, 2009]. This definition excludes other abnormalities of CSF dynamics, such as benign intracranial hypertension, in which the ventricles are not enlarged. It does not specify the source of production or absorption of CSF nor does it presuppose the mechanisms inherent in ventricular distension.
Classification
A number of classification systems for hydrocephalus have been suggested [Mori, 1995; Boaz and Edwards-Brown, 1999; Beni-Adani et al., 2006; Oi and Di Rocco, 2006; Rekate, 2009]. These include the following types of hydrocephalus:






The terms compensated and uncompensated hydrocephalus generally refer to whether an increase in ventricular size is associated with evidence of raised intracranial pressure. In compensated, or arrested, hydrocephalus, a gradual increase in ventricular size stabilizes by reaching a new equilibrium and the patient has no symptoms or signs of raised intracranial pressure. In contrast, uncompensated hydrocephalus is associated with clinical symptoms and signs of raised intracranial pressure and usually with progressive dilatation of the ventricles. This is the clinical situation where treatment is indicated. No classification is completely adequate for hydrocephalus at all ages. The classification of hydrocephalus into communicating or noncommunicating dates back to the early 1900s and is based on Walter Dandy’s experimental studies into the pathophysiology of hydrocephalus [Richards, 1990].
Hydrocephalus is also categorized as congenital, which is present at birth and often associated with developmental defects; and acquired, which occurs after development of the brain and ventricles [Mori, 1995; Chahlavi et al., 2001]. Hydrocephalus has also been classified based on the stage of development at the time that the ventricles became dilated [Oi and Di Rocco, 2006]. The various subtypes of fetal hydrocephalus are classified according to the mechanism of obstruction to the flow of CSF to include:
This classification is cross-referenced to the stage of fetal development (e.g., neuronal maturation, cell migration); it may prove useful in deciding when treatment may be futile if beyond the legal period for terminating a pregnancy, and in identifying potential candidates for early delivery or fetal surgery [Rekate, 2009].
Extraventricular obstructive hydrocephalus is now recognized to represent, almost universally, benign pericerebral collections of infancy that are usually familial, resolve with time, and almost never require treatment [Drake, 2008].
Benign extra-axial collections of infancy involve abnormal enlargement of the head and excessively large subarachnoid space. In infants whose neurological development is normal and in whom the enlargement of the subarachnoid space resolves by 24 months of age, no further work-up or treatment is required. In infants whose neurological development is abnormal or in whom the enlargement of the subarachnoid space does not resolve by 24 months of age, further investigations may be warranted. Several genetic conditions, such as certain mucopolysaccharidoses, achondroplasia, Sotos’ syndrome, and glutaric aciduria type I, can feature enlargement of the subarachnoid spaces as either an early or an associated finding on neuroimaging. In the case of the mucopolysaccharidoses, enlargement of the subarachnoid spaces may be a direct consequence of impairment of CSF absorption by the storage material. Thus, enlargement of the subarachnoid spaces may be an important clue to an early genetic diagnosis, which is important, as therapeutic options exist for many forms of mucopolysaccharidosis and for glutaric aciduria type I [Paciorkowski and Greenstein, 2007].
Epidemiology
In newborns, the overall incidence of hydrocephalus ranges from 0.3 to 4 per 1000 live births. Occurring as a single congenital disorder, the incidence of hydrocephalus has been reported as 0.9–1.5 per 1000 births [Milhorat, 1972; Serlo et al., 1986; El Awad, 1992; Blackburn and Fineman, 1994; Fernell and Hagberg, 1998]. It is estimated that approximately 125,000 persons are living with ventricular shunts and that 33,000 shunts are placed annually in the United States [Bondurant and Jimenez, 1995].
The incidence of pediatric hydrocephalus has declined in many developed countries [Drake, 2008]. Antenatal screening, genetic testing, and pregnancy termination have reduced the incidence of congenital malformations of the brain that cause hydrocephalus. The incidence of open neural tube defects has also decreased precipitously as a result of maternal folate supplementation, antenatal screening, and termination of pregnancy based on superior antenatal imaging with ultrasound and magnetic resonance imaging (MRI). The incidence of CSF shunting in open neural tube defects, formerly reported to be as high as 90 percent, has also declined, possibly as a result of a general, more conservative approach, and also the selection of lower-grade lesions for delivery with a lower need for shunting [Tulipan et al., 2003; Chakraborty et al., 2008]. The incidence of intraventricular hemorrhage (IVH) has also decreased as a result of better perinatal management of prematurity, and, as such, one of the major complications of post-hemorrhagic hydrocephalus (see Chapter 19) [Fernell and Hagberg, 1998].
CSF Production, Circulation, and Absorption
CSF is produced by two mechanisms: Most of the CSF (50–80 percent) is thought to be secreted by the choroid plexus within the cerebral ventricles. Extrachoroidal CSF production in subarachnoid sites and by way of a transependymal route has also been documented. About 20 percent or more of CSF is derived from brain extracellular fluid created as a byproduct of cerebral metabolism [Bering, 1962; Fishman, 1980; Rekate, 1997]. Normally, rates of production (0.35 mL per minute or approximately 400–500 mL/day) and absorption of CSF are equal. Total CSF volume is 65–140 mL in children, and 90–150 mL in adults. In one study of premature infants born at gestational age 32 ± 1.6 wk, CSF volume was estimated to be 23.33 ± 9.6 mL at birth and 40.66 ± 21.23 mL at term equivalent. Full-term infants have a CSF volume of 32.4 ± 11.1 mL at birth [Zacharia et al., 2006].
The process of CSF formation by the choroid plexus includes plasma ultrafiltration and secretion. Secretion, an energy-dependent process, is initiated by hydrostatic pressure in the choroidal capillaries and by active transport of sodium. The enzymes sodium-potassium adenosine triphosphatase (ATPase) and carbonic anhydrase partly regulate CSF secretion [Fishman, 1980]. CSF production has been reported to remain constant across the normal intracranial pressure range [Pollay, 1977], with CSF production decreasing when intracranial pressure approaches mean arterial pressure. There have been reports, however, of downregulation of CSF production in patients with chronic hydrocephalus [Silverberg et al., 2002].
By contrast, the process of CSF reabsorption is not an energy-dependent process [Rekate, 1997]. After formation, CSF exits from the lateral ventricles through the foramina of Monro into the third ventricle (Figure 27-1). CSF then traverses the aqueduct of Sylvius into the fourth ventricle, leaving though the foramina of Lushka and foramen of Magendie into the cisterna magna, from where it flows through the subarachnoid space around the cerebral hemispheres. Information gained from MRI analysis of CSF movement demonstrates pulsatile to-and-fro motion of CSF within the lateral ventricles, produced from a brain-pumping motion that ejects the CSF and causes a net downward flow [Feinberg and Mark, 1987].
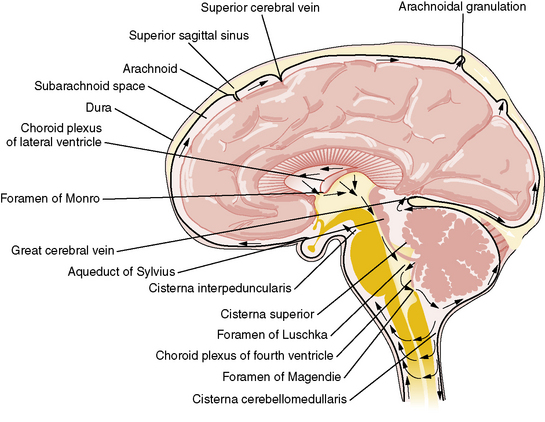
Historically, it has been held that CSF is absorbed into the vascular system mainly through the arachnoid villi within the arachnoid granulations covering the brain and spinal cord leptomeninges [Alksne and Lovings, 1972; Welch, 1975]. This process is thought to be passive and not energy-dependent. A layer of endothelium within the arachnoid villi separates the subarachnoid CSF space from the vascular system. Water and electrolytes pass freely across these arachnoid membranes. There is normally 5–7 mmHg difference in pressure between the dural venous sinuses and the subarachnoid space. This is presumed to be the hydrostatic force behind the absorption of CSF. Larger proteins and macromolecules cannot pass through intercellular junctions but are selectively transported across the cytoplasm of endothelial cells by an active process involving micropinocytosis [Welch, 1975]. Increased absorption through the arachnoid villi protects the brain from transient increases in intracranial pressure [Mann et al., 1978]. Newborn infants do not have visible arachnoid granulations, suggesting that the maximum capacity for reabsorption is less than in the adult, or that CSF is absorbed by different mechanisms in the neonate.
For some time, arachnoid granulations were thought to be the only CSF absorption pathway; however, other mechanisms have been identified, with evidence emerging that arachnoid granulations have only a secondary role. Olfactory nerves, the cribriform plate, and nasal lymphatics have been identified as important sites for CSF absorption [Johnston and Papaiconomou, 2002; Johnston, 2003; Johnston et al., 2004].
Absorption of CSF across brain tissue into capillaries has also been proposed [Greitz, 2004]. According to this theory, the distending force in the production of chronic hydrocephalus is an increased systolic pulse pressure in the brain tissue, due to decreased intracranial compliance.
Etiology and Pathophysiology
Hydrocephalus can be a symptom of a large number of disorders, and a list of conditions in which it has been reported is summarized in Box 27-1. It is associated with tumors and infections, and may be a complication of prematurity and trauma [Renier et al., 1988; Schrander-Stumpel and Fryns, 1998; Chi et al., 2005]. It is also seen in apparent isolation. High-resolution MRI of postnatal life has provided clues to the etiology of hydrocephalus, which in the past would have been labeled as idiopathic; some of these include IVH (Figure 27-2), aqueductal stenosis (Figure 27-3), and migrational abnormalities [Drake, 2008].
Box 27-1 Differential Diagnosis of Hydrocephalus



































































CRASH, corpus callosum agenesis, retardation, adducted thumbs, spastic paraparesis, hydrocephalus; VACTERL, vertebral anomalies, anal atresia, cardiac defect, tracheoesophageal fistula with esophageal atresia, renal abnormalities, limb abnormalities.
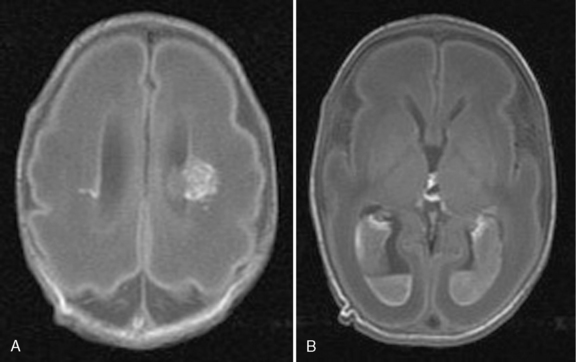
The etiologies of hydrocephalus in one series of pediatric patients are shown in Table 27-1 [Drake et al., 1998]. Hydrocephalus is due to either abnormal CSF reabsorption, or flow, or, rarely, overproduction. The main situation in which CSF production is increased enough to cause hydrocephalus is the presence of a choroid plexus papilloma. These tumors contain functional choroid epithelium and can produce very large amounts of CSF. However, even in the latter case, reabsorption is probably defective, as normal individuals can usually tolerate the elevated CSF production rate of these tumors. CSF accumulation, in turn, leads to raised intracranial pressure.
Table 27-1 Causes of Hydrocephalus
| Causes | Percentage |
|---|---|
| Intraventricular hemorrhage | 24.1 |
| Myelomeningocele | 21.2 |
| Tumor | 9.0 |
| Aqueductal stenosis | 7.0 |
| Infection | 5.2 |
| Head injury | 1.5 |
| Other | 11.3 |
| Unknown | 11.0 |
| Two or more causes | 8.7 |
(Modified from Drake JM, et al: Randomized trial of cerebrospinal fluid shunt valve design in pediatric hydrocephalus. Neurosurgery 43[2]:294–303, 1998; discussion 303–295.)
Congenital Causes in Infants and Children
Approximately 55 percent of all cases of hydrocephalus are congenital. Primary aqueductal stenosis accounts for approximately 5 percent of congenital hydrocephalus, whereas aqueductal stenosis secondary to neoplasm, infection, or hemorrhage accounts for another 5 percent [Chi et al., 2005] . Primary aqueductal stenosis usually presents in infancy. Its morphology may be that of “forking” of the aqueduct, an aqueductal septum, “true” narrowing of the aqueduct, or X-linked aqueductal stenosis. Bicker–Adams syndrome is an X-linked hydrocephalus accounting for 7 percent of cases in males. It is characterized by stenosis of the aqueduct of Sylvius, severe mental retardation, and, in 50 percent, an adduction-flexion deformity of the thumb. Secondary aqueductal stenosis is due to gliosis secondary to intrauterine infection or germinal matrix hemorrhage [Hill and Rozdilsky, 1984].
Anatomical malformations frequently observed with idiopathic congenital hydrocephalus are associated with abnormalities of hindbrain development (see Chapter 24), and include Chiari malformations, Dandy–Walker malformation (DWM), and others [Stoll et al., 1992; Schrander-Stumpel and Fryns, 1998]. DWM is associated with atresia of the foramina of Luschka and Magendie, and affects 2–4 percent of newborns with hydrocephalus. About 50 percent of all patients with DWM develop hydrocephalus. The dilated fourth ventricle does not communicate effectively with the subarachnoid space. In patients with Chiari malformations, hydrocephalus may occur, with fourth ventricle outlet obstruction in Chiari type 1 malformation; it is commonly associated with myelomeningocele in the Chiari type 2 malformation. Hydrocephalus occurs in approximately 80–90 percent of patients with myelomeningocele; of these cases, 50 percent are obvious at birth [Tuli et al., 2003; Tulipan et al., 2003].
Acquired Causes in Infants and Children
Post-hemorrhagic hydrocephalus (PHH) occurs following IVH and can be related to prematurity, head injury, or rupture of a vascular malformation. Communicating hydrocephalus post-subarachnoid hemorrhage is more common in adults and is rarely seen in children. Over the past two decades, there has been remarkable improvement in the survival of extremely low birth weight infants; however, the most immature of these infants remain at increased risk for neonatal complications that potentially affect long-term neurodevelopmental outcome, including IVH (see Chapter 19). The risk for severe IVH varies inversely with gestational age, with an overall incidence of 7–23 percent [Lemons et al., 2001; Ment et al., 2005; Wilson-Costello et al., 2005]. Approximately one-third of extremely low birth weight infants with an IVH develop PHH, 15 percent of whom will require shunt insertion [Dykes et al., 1989; de Vries et al., 2002; Kazan et al., 2005].
Hydrocephalus after IVH is usually ascribed to fibrosing arachnoiditis, meningeal fibrosis, and subependymal gliosis, which impair flow and resorption of CSF. Recent experimental studies have suggested that acute parenchymal compression and ischemic damage, and increased parenchymal and perivascular deposition of extracellular matrix proteins – probably due, at least partly, to upregulation of transforming growth factor-beta (TGF-β) – are further important contributors to the development of the hydrocephalus. IVH is associated with damage to periventricular white matter and the damage is exacerbated by the development of hydrocephalus; combinations of pressure, distortion, ischemia, inflammation, and free radical-mediated injury are probably responsible [Cherian et al., 2004].
Clinical Characteristics
The clinical features of hydrocephalus depend on the age of the child at presentation and the time of onset in relation to closure of the cranial sutures. With the current advances in antenatal monitoring, the majority of congenital cases of hydrocephalus are diagnosed early (Figure 27-4), allowing for planned cesarian delivery in the moderate to severe cases where cephalopelvic disproportion is expected.

Fig. 27-4 Antenatal MRI showing fetal hydrocephalus.
Symmetrical enlargement of the ventricular system is readily seen in the brain of this fetus.
Genetics
Although commonly considered a single disorder, hydrocephalus is a collection of heterogeneous complex and multifactorial disorders. A growing body of evidence suggests that genetic factors play a major role in its pathogenesis [Zhang et al., 2006].
Congenital hydrocephalus may occur alone (nonsyndromic) or as part of a syndrome with other anomalies (syndromic). It is estimated that about 40 percent of individuals with hydrocephalus have a genetic etiology [Haverkamp et al., 1999]. The isolated (nonsyndromic) form of congenital hydrocephalus is a primary and major phenotype caused by a specific faulty gene. In syndromic forms, it is difficult to define the defective gene because of the association with other anomalies. A discussion of the genetics of all syndromic forms of hydrocephalus is beyond the scope of this chapter and is reviewed elsewhere in this section on developmental malformations. This chapter will focus mainly on the genetic disorders associated with isolated forms of hydrocephalus.
Autosomal-recessive, autosomal-dominant, X-linked recessive [Castro-Gago et al., 1996] and X-linked dominant [Ferlini et al., 1995] forms of hydrocephalus are recognized (Table 27-2).
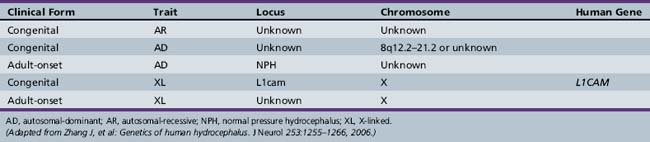
At least 43 gene mutations linked to hereditary hydrocephalus have been identified in animal models and humans. To date, nine genes associated with hydrocephalus have been identified in animal models, whereas only one such gene has been identified in humans: the hydrocephalus (X-linked) gene [Haverkamp et al., 1999]. X-linked hydrocephalus (HSAS1, OMIM) occurs in approximately 5–15 percent of congenital cases in which a genetic etiology is determined [Halliday et al., 1986; Haverkamp et al., 1999]. The gene responsible for X-linked human congenital hydrocephalus is at Xq28, encoding for L1CAM (L1 cell adhesion molecule) [Jouet et al., 1993]. Mutations are distributed over the functional protein domains. The exact mechanisms by which these mutations cause a loss of L1 protein function are still under investigation. L1CAM belongs to the immunoglobulin superfamily of neural cell adhesion molecules that is expressed in neurons and Schwann cells and appears to be essential for brain development and function. It plays a role in cell adhesion, axon growth and path-finding, and neuronal migration and myelination [Wong et al., 1995]. One of the possible mechanisms leading to the pathogenesis of hydrocephalus is the disruption of neural cell membrane proteins that play an important function during brain development.
The L1 disorders were initially described as different entities:
During the 1990s, it became clear that these disorders resulted from a mutation in a single gene, L1CAM [Fransen et al., 1995]. Some investigators have suggested that these separately named conditions be combined under the acronym CRASH (corpus callosum agenesis, retardation, adducted thumbs, shuffling gait, and hydrocephalus). The clinical phenotype is variable, even within a single family, but some genotype–phenotype correlations have been made [Fransen et al., 1998]. Hirschsprung’s disease (HSCR) is characterized by the absence of ganglion cells and the presence of hypertrophic nerve trunks in the distal bowel. There have been several reports of patients with X-linked hydrocephalus and HSCR with a mutation in the L1CAM gene. Decreased L1CAM may therefore be a modifying factor in the development of HSCR [Okamoto et al., 2004]. Congenital aqueductal stenosis can also be inherited as an autosomal-recessive disorder (OMIM 236635) [Lapunzina et al., 2002].
In general, the recurrence risk for congenital hydrocephalus excluding X-linked hydrocephalus is low. Empiric risk rates range from <1 to 4 percent [Burton, 1979], indicating the rarity of autosomal-recessive congenital hydrocephalus [Halliday et al., 1986; Chow et al., 1990; Haverkamp et al., 1999]. However, multiple human kindreds have been reported [Halliday et al., 1986; Teebi and Naguib, 1988; Haverkamp et al., 1999; Zhang et al., 2006]. The loci or genes for human autosomal-recessive congenital hydrocephalus have not yet been identified, but there is at least one locus for this trait. Furthermore, since there is heterogeneity among clinical phenotypes, there may be more genetic loci in human autosomal-recessive congenital hydrocephalus.
Two kindreds in which congenital hydrocephalus was transmitted in an autosomal-dominant fashion have been reported. One was associated with aqueductal stenosis but was not associated with mental retardation or pyramidal tract dysfunction, which is in contrast to X-linked or recessive congenital hydrocephalus with HSAS, in which these abnormalities are common [Verhagen et al., 1998]. The other kindred in which hydrocephalus developed had an 8q12.2–q21.2 microdeletion. This trait was also transmitted in an autosomal-dominant pattern [Vincent et al., 1994].
Hydrocephalus has been observed in many mammals [Zhang et al., 2006]. Animal hydrocephalus models have many histopathological similarities to humans and can be used to understand the genetics and pathogenesis of these disorders. It has been well documented that the majority of cases of congenital hydrocephalus in animal models occur on a genetic basis with specific mapping and identification of different loci. The development and progression of congenital hydrocephalus constitute a dynamic process that is not yet well understood. It is probably the consequence of abnormal brain development and perturbed cellular function, which emphasizes the important roles that congenital hydrocephalus genes play during brain development. It is thought that it may develop at an important and specific embryonic time period of neural stem-cell proliferation and differentiation [Zhang et al., 2006].
Hydrocephalus may also be caused by alterations in ependymal cell function [Takano et al., 1993; Wagner et al., 2003]. The protein of the axonemal heavy chain 5 gene (Mdnah5), dynein, is specifically expressed in ependymal cells, and is essential for ultrastructural and functional integrity of ependymal cilia. In Mdnah5-mutant mice, lack of ependymal flow causes closure of the aqueduct and subsequent formation of triventricular hydrocephalus during early postnatal brain development. The higher incidence of aqueductal stenosis and hydrocephalus formation in patients with disorders associated with ciliary defects supports the relevance of this novel mechanism in humans [Ibanez-Tallon et al., 2004].
Hydrocephalus may be caused by changes in the development and function of mesenchymal cells. In murine embryonic brain, the Msx1 gene is expressed along the dorsal midline. This regulatory gene is involved in epithelio-mesenchymal interactions in limb formation and organogenesis. In homozygous Msx1 mutants, there is absence or malformation of the posterior commissure and of the subcommissural organ, collapse of the cerebral aqueduct, and hydrocephalus. About one-third of the heterozygous mutants also develop hydrocephalus, suggesting that the phenotype may be determined by the Msx1 gene dosage during a critical developmental period [Ramos et al., 2004]. An example of this is the autosomal-recessive congenital hydrocephalus mouse model in which a truncated protein lacking the DNA-binding domain of the forkhead/winged helix gene, Mf1, was generated. Mesenchymal cells from Mf1lacZ embryos differentiate poorly into cartilage in micromass culture, and the differentiation of arachnoid cells in meninges of the mutant mice also was abnormal. Human patients with deletions in the region containing human Mf1 homolog FREAC3 were also found to develop multiple developmental disorders, including hydrocephalus [Kume et al., 1998].
Another mechanism for development of hydrocephalus may be associated with changes in growth factor signaling [Fukumitsu et al., 2000]. In mouse models, severe hydrocephalus has been observed in transgenic mice overexpressing TGF-β1, an important cytokine and growth-signaling molecule, in astrocytes [Galbreath et al., 1995]. In mouse models, fibroblast growth factor-2 (FGF-2) seems to play a predominant role in the proliferation of neuronal precursors and in neuronal differentiation in the developing cerebral cortex, even at relatively late stages of brain neurogenesis [Ohmiya et al., 2001].
Hydrocephalus may also be caused by disruption of the extracellular matrix (ECM). In the TGF-β1 overexpression mouse model, the changing expressions of a remodeling protein, matrix metalloproteinase-9 (MMP-9), and its specific inhibitor, tissue inhibitor of metalloproteinases-1 (TIMP-1), were found to be important factors in the spontaneous development of hydrocephalus by altering the ECM environment [Crews et al., 2004]. Furthermore, increased expression of cytokines such as TGF-β1 might reciprocally play an important role by disrupting vascular ECM remodeling, promoting hemorrhage and altering reabsorption of CSF. In another mouse model, ablation of the non-muscle myosin heavy chain II-B (NMHC-B) results in severe hydrocephalus with enlargement of the lateral and third ventricles. These defects may be caused by abnormalities in the cell adhesive properties of neuroepithelial cells, and suggest that NMHC-B is essential for early and late developmental processes in the mammalian brain [Tullio et al., 2001].
Other genetic mutations identified in animal models of hydrocephalus include those coding α-SNAP protein, which is essential for apical protein localization and cell fate determination in neuroepithelial cells [Chae et al., 2004]. α-SNAP plays a key role in a wide variety of membrane fusion events in eukaryotic cells, a function which is required for transport of molecules to inter- and intracellular compartments and for intercellular communication.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


