CHAPTER 53 Antiepileptic Medications
Principles of Clinical Use
This chapter presents a systematic review of the clinical pharmacology of each of the main established AEDs and the newer AEDs, including their pharmacokinetics, interactions, dosages, efficacy profile, and safety profile. The pharmacokinetic properties, as well as doses and therapeutic ranges of the AEDs to be discussed, are summarized in Table 53-1. The principles of drug treatment of epilepsy are discussed, such as the decision to initiate long-term prophylactic drug treatment, the sequence of drug choices for various seizure types or syndromes (Table 53-2), initiation and monitoring of antiepileptic therapy, and discontinuation of treatment.
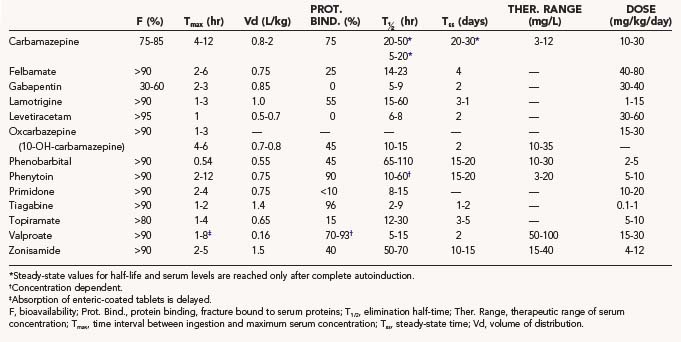
TABLE 53-2 Place of Newer Antiepileptic Drugs in the Treatment Sequence of Seizures and Epileptic Syndromes in Children
| Partial Seizures with or without Secondary Generalization | |
| First choice | Oxcarbazepine, carbamazepine, levetiracetam |
| Second choice | Lamotrigine, valproate, gabapentin |
| Third choice | Topiramate, zonisamide, phenytoin, phenobarbital, primidone |
| Consider | Pregabalin, tiagabine, benzodiazepine, acetazolamide |
| Generalized Tonic-Clonic Seizures | |
| First choice | Valproate, levetiracetam, lamotrigine |
| Second choice | (Ox)carbamazepine, topiramate, phenytoin |
| Third choice | Zonisamide, phenobarbital, primidone |
| Absence Seizures | |
| Before 10 years of age | |
| First choice | Ethosuximide, valproate |
| Second choice | Lamotrigine |
| Consider | Methsuximide, benzodiazepine, levetiracetam, topiramate, zonisamide, acetazolamide |
| After 10 years of age | |
| First choice | Valproate |
| Second choice | Lamotrigine |
| Third choice | Ethosuximide, methsuximide, levetiracetam, topiramate, zonisamide, benzodiazepine, acetazolamide |
| Juvenile Myoclonic Epilepsy | |
| First choice | Valproate |
| Second choice | Levetiracetam, lamotrigine, topiramate, clonazepam |
| Third choice | Zonisamide, phenobarbital, primidone |
| Lennox-Gastaut and Related Syndromes | |
| First choice | Topiramate, lamotrigine |
| Second choice | Valproate |
| Third choice | Ketogenic diet, felbamate, zonisamide, rufinamide, benzodiazepine, phenobarbital |
| Consider | Ethosuximide, methsuximide, levetiracetam, steroids |
| Infantile Spasms | |
| First choice | Adrenocorticotropic hormone, vigabatrin |
| Second choice | Valproate, topiramate, lamotrigine, zonisamide, benzodiazepine, ketogenic diet |
| Consider | Pyridoxine, levetiracetam, felbamate |
| Benign Epilepsy with Centrotemporal Spikes | |
| First choice | Sulthiame, gabapentin |
| Second choice | Valproate, levetiracetam |
| Consider | Lamotrigine, topiramate, zonisamide, pregabalin |
Clinical Pharmacology of Antiepileptic Drugs
Older Antiepileptic Drugs
Phenytoin
The use of phenytoin (PHT) has decreased, but it is still a widely used AED. In addition to its good efficacy against convulsive seizures and decades of experience with a good safety profile, PHT can be loaded rapidly without the need to titrate the dose slowly. This property is particularly valuable in neurosurgical practice. The volume of distribution of PHT is about 0.75 L/kg. A dose of 7.5 mg/kg given intravenously raises the level by 10 mg/L. Elimination of PHT is unique among AEDs because it is saturable at therapeutic concentrations1 in all age groups.2,3 This results in a nonlinear relationship between maintenance doses and steady-state concentrations. Especially in the upper therapeutic range, small increases in dosage can cause relatively large increases in levels. PHT does not have an elimination half-life because the time for the level to decrease by 50% becomes longer at higher levels. Steady-state levels are reached only after 2 to 3 weeks of a stable maintenance dose. To achieve average levels of about 15 mg/L, adult patients must usually take 5 to 6 mg/kg per day, which corresponds to 350 to 450 mg/day. The common dose of 300 mg/day often results in levels of 10 mg/L or less. PHT is a potent enzyme inducer and lowers the level of many other drugs. This affects other AEDs, such as carbamazepine, valproate, felbamate, lamotrigine, topiramate, zonisamide, and tiagabine, as well as many other drugs, including warfarin, oral contraceptives, and cyclosporine. PHT is highly protein bound and is displaced from serum proteins by valproate. Such displacement increases the free fraction of PHT and makes total serum levels unreliable.
The spectrum of activity of PHT includes partial seizures (simple or complex without or with secondary generalization), generalized convulsive seizures, status epilepticus,4 and neonatal seizures.5 Intravenous administration of PHT can cause bradyarrhythmia and hypotension, as well as skin necrosis.6 This local irritation in particular can be avoided by using the prodrug phosphenytoin instead of PHT for intravenous administration. The rate of administration of phosphenytoin is up to 150 mg/min (or 3 mg/kg per minute) instead of 50 mg/min (or 1 mg/kg per minute) for PHT.
The dose-related central nervous system side effects of PHT are nystagmus, ataxia, and lethargy. PHT can cause various forms of hypersensitivity reactions,7 as well as a hypersensitivity syndrome.8 Chronic or delayed adverse effects include gingival hyperplasia, hirsutism, peripheral neuropathy, and bone demineralization secondary to reduced vitamin D levels.
Carbamazepine
Carbamazepine (CBZ) is gradually being displaced from its exclusive place as the drug of first choice for partial and secondarily generalized seizures. The elimination kinetics of CBZ is linear.9 The characteristic feature of CBZ elimination is autoinduction of its metabolism,10 which results in an increase in CBZ clearance during the first weeks of treatment unless the patient already is taking another enzyme-inducing drug, such as phenytoin, phenobarbital, or primidone. Accordingly, the elimination half-life of CBZ decreases from about 36 hours to 10 to 20 hours.11 The practical consequence is that the dose of CBZ must be increased progressively during the first 3 to 4 weeks of treatment from 100 to 200 mg/day to 600 to 800 mg/day. Further increases in dosage may be needed and tolerated. Because of the relatively short half-life after induction, the regular CBZ preparations are best taken three times a day. Slow-release preparations are taken every 12 hours. CBZ is involved in many pharmacokinetic interactions. Similar to phenytoin, it is an enzyme inducer (see earlier). Other enzyme-inducing drugs, such as phenytoin, phenobarbital, and primidone, accelerate CBZ metabolism to a degree that exceeds CBZ autoinduction.
CBZ is effective against partial seizures without or with secondary generalization, as well as against generalized tonic-clonic seizures. Against partial and secondarily generalized seizures, phenytoin, CBZ, phenobarbital, and primidone are about equally effective in terms of seizure control.12 There is no parenteral preparation for CBZ. Dose-related central nervous system toxicity is the most common side effect. This toxicity may subside with time, can be minimized by careful titration, and is closely related to CBZ serum levels.13 Other side effects include neutropenia and rare, severe blood dyscrasias,14 hyponatremia, movement disorders, allergic rashes, and hypersensitivity syndrome.
Valproate
Valproate (VPA) is unique among the older AEDs because of its broad spectrum of activity against various seizure types. Absorption from enteric-coated VPA tablets can be delayed by several hours, but once it begins, it is rapid, as opposed to the slow release from extended-release tablets. Other oral preparations exhibit rapid and early absorption, except for enteric-coated sprinkles, which have an intermediate absorption pattern. VPA is highly bound to serum proteins and tends to displace other drugs, such as phenytoin. The elimination half-life of VPA varies as a function of comedication. In adults, the half-life is 13 to 16 hours in the absence of inducing drugs15 and 9 hours in induced patients.16 In addition to displacement from serum proteins, VPA is involved in two types of pharmacokinetic interactions: its metabolism is accelerated by inducing drugs such as phenytoin, carbamazepine, phenobarbital, and primidone, and VPA itself can prolong the elimination (and raise the levels) of other drugs, such as phenobarbital, ethosuximide, lamotrigine, and felbamate. The initial target dose of VPA is 15 mg/kg per day, which can be attained within a few days. Higher doses of 60 mg/kg per day or more may be necessary in certain patients, especially in children and those taking inducing drugs.
VPA has a broad spectrum of activity. In addition to being effective against partial seizures,17 VPA is highly effective against absence seizures, generalized tonic-clonic seizures, and myoclonic seizures. It is a drug of first choice in patients with primary (idiopathic) generalized epilepsies. It can be helpful in the treatment of infantile spasms18 and Lennox-Gastaut syndrome. VPA has several side effects that affect different systems and are of variable severity. Mild side effects include transient hair loss and dose-related tremor. VPA is not sedative, but drowsiness and lethargy may appear in some patients at levels around 100 mg/L, as well as idiosyncratic stuporous states at therapeutic levels.19 Gastrointestinal upset is less common with enteric-coated tablets. Fatal hepatotoxicity20 and pancreatitis21 are the most serious complications of VPA treatment. Thrombocytopenia, in conjunction with other VPA-mediated disturbances of hemostasis, such as impaired platelet function, fibrinogen depletion, and coagulation factor deficiencies,22 may cause excessive bleeding. The common practice of withdrawing VPA before elective surgery is recommended, although reports have found no objective evidence of excessive operative bleeding in neurosurgical patients maintained on VPA.23,24 In women of childbearing age, concerns associated with VPA treatment include not only an increased risk for neural tube defects in the fetus but also an increased risk for polycystic ovaries and metabolic and endocrine disturbances.25
Phenobarbital and Primidone
The use of phenobarbital (PB) and primidone (PRM) for the treatment of seizures has declined steadily because of their central nervous system side effects. PB and PRM produce more sedative and behavioral side effects than most other AEDs do, but they have relatively little systemic toxicity. PB has excellent pharmacokinetic properties, can be administered intravenously and intramuscularly, is effective in patients with status epilepticus, and is inexpensive. The volume of distribution of PB is 0.55 L/kg, with an elimination half-life averaging 80 to 100 hours in adults and newborns and shorter in infants and children. Maintenance doses range from 2 to 5 mg/kg per day. Although treatment with PRM results in the accumulation of significant levels of PB, PRM has independent pharmacologic activity and probably is not just a prodrug. PRM itself has a much shorter half-life than PB. Daily dosage requirements of PRM are about five times higher than those of PB. As an enzyme inducer, PB causes pharmacokinetic interactions that are shared by other enzyme-inducing drugs and by PRM because of the derived PB. Other enzyme-inducing drugs, in particular phenytoin,26 accelerate the conversion of PRM to PB, thereby increasing the PB-to-PRM serum level ratio.
PB and PRM are as effective against partial and secondarily generalized seizures as carbamazepine and phenytoin but were found to be associated with more treatment failures because of mostly early central nervous system side effects.12 PB can be used for the treatment of status epilepticus and neonatal seizures, as well as for the prophylaxis of febrile seizures. In addition to the well-known sedative and behavioral side effects, PB and PRM can cause allergic reactions. Use over many years may be associated with connective tissue disorders, such as Dupuytren’s contracture and frozen shoulder.
Newer Antiepileptic Drugs
Felbamate
Because of potentially serious side effects, felbamate (FBM) is currently used only in special circumstances. FBM is involved in multiple pharmacokinetic interactions; its levels are decreased by enzyme-inducing drugs, and it raises levels of phenytoin and valproate. The recommended initial dose of FBM is 1200 mg/day (15 mg/kg per day) during the first week. This dose can be doubled at the beginning of the second week and tripled at the beginning of the third week. It is prudent to reduce the dose of other AEDs by about a third when FBM is introduced. In double-blind studies, FBM was shown to be effective against partial onset seizures,27,28 as well as in the treatment of Lennox-Gastaut syndrome.29 Uncontrolled reports have suggested efficacy of FBM against absence seizures, juvenile myoclonic epilepsy, and infantile spasms.
The main common side effects of FBM have been nausea and vomiting, anorexia and weight loss, somnolence, and insomnia. Within 1 year after its marketing, it became evident that FBM was associated with a relatively high incidence of potentially fatal aplastic anemia30 and hepatic necrosis.31 Currently, the main indication for FBM is as a drug of third choice for the treatment of Lennox-Gastaut syndrome and focal onset seizures.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


